
“Legacy” medical devices and IVDs under EU legislation
The new EU regulations on medical devices and in-vitro diagnostic devices allow devices CE-marked per the former EU directives to still be placed on the market under certain conditions. Such devices are referred to as “legacy devices”.
If you are uncertain about the legacy qualification of your medical device or in-vitro diagnostic device (IVD), or you wish to know more about how legacy devices are regulated under the EU MDR or IVDR, read on.
Replaces the version of April 2023.
Key takeaways:
- Not all devices qualify as “legacy”. If your Class I device or self-declared IVD would not require the involvement of a Notified Body under the EU MDR or IVDR, it should already be in compliance with the new Regulation. If you need help with compliance, reach out to us.
- Changes in device design or intended purpose trigger the loss of the “legacy” status. Make sure you are familiar with the restrictions and that your change management process is aligned.
- Transition periods for legacy devices have changed both under the EU MDR and the IVDR. Check the latest timelines and applicable conditions in this article. And do not forget that, for foreign manufacturers, all legacy devices sold in Switzerland require a Swiss Authorised Representative.
Content:
When does a CE-marked device (medical device or IVD) qualify as legacy device?
Legacy devices under the EU MDR and IVDR are those devices allowed to be placed on the market after the date of application of the corresponding regulation (i.e. 26 May 2021 for the EU MDR and 26 May 2022 for the IVDR), subject to the transitional provisions in EU MDR Article 120(3) and Article 110(3), respectively.
The following devices may qualify as “legacy”, provided the conditions in EU MDR Article 120 or IVDR Article110 are fulfilled for each case:
- All medical devices that still have a valid Notified Body certificate (issued in accordance with the former MDD, AIMDD or IVDD),
- Medical devices only (i.e. excluding IVDs) with a Notified Body certificate that has expired before 20 March 2023, and
- Those devices that did not require the involvement of a Notified Body for CE marking under the former Directives (thus, with no certificate) but would now require so under the EU MDR or IVDR qualify as legacy devices.
This means that the following categories are NOT considered legacy devices:
- Class I devices (non-sterile and without measuring function) under the MDD that would remain Class I devices under the EU MDR as long as they do not have a measuring function (i.e. Class Im), are not sterile (i.e. Class Is) and are not surgically reusable devices (i.e. Class Ir).
- Self-declared IVDs (non-sterile) under the IVDD that would correspond to non-sterile Class A IVDs under the IVDR.
Note that devices that were last placed on the market before the date of application of the corresponding regulation, i.e. 26 May 2021 for the EU MDR and 26 May 2022 for the IVDR, are considered to be “old devices”, not legacy devices. Despite the fact that old devices on the market may remain in use in the field for a long time (e.g. surgical equipment, laboratory centrifuges for IVD testing), they are not subject to the EU MDR or IVDR requirements, with the exception of serious incident reporting. See section What EU MDR or IVDR requirements apply to legacy devices? for details.
There are three situations where legacy devices can lose their legacy status:
- By implementing significant changes to the device design or intended purpose. See section What is a “significant change” under the EU MDR and IVDR?
- By reaching the end of the corresponding transitional period in the EU MDR or IVDR. See section What transitional periods and conditions apply to legacy devices?
- Due to expiration of the Notified Body certificate issued under the MDD/AIMDD or IVDD, without fulfilling the conditions to benefit from the transitional periods in the EU MDR or IVDR. See section What transitional periods and conditions apply to legacy devices?
What about Custom-made devices?
According to the definition in EU MDR Article 2(3), custom-made device means:
“any device specifically made in accordance with a written prescription of any person authorised by national law by virtue of that person’s professional qualifications which gives, under that person’s responsibility specific design characteristics, and is intended for the sole use of a particular patient exclusively to meet their individual conditions and needs”
In summary this means that custom-made devices are medical devices that are given design properties to match a specific patient’s needs (i.e. not mass produced). A classic example of a custom-made device is a prosthetic limb. The device is made on prescription issued by an authorised person (i.e. a specialist doctor such as an orthopedist).
“Manufacturers of custom-made devices must follow EU MDR requirements, even if no certification is required”
Custom-made devices were not covered by the initial transitional provisions in EU MDR Article 120. Thus, they did not qualify as legacy devices and had to comply with EU MDR requirements since 26 May 2021. However, the new amending Regulation (EU) 2023/607 that extends the transition timelines in the EU MDR has introduced a “grace period” for Class III implantable custom-made devices, which would need Quality Management System (QMS) certification by a Notified Body, per EU MDR Article 52(8). According to the new EU MDR Article 120(3)(f), the certification can be postponed until 26 May 2026, provided the manufacturer has lodged a formal application with a Notified Body by 26 May 2024 and signed the corresponding written agreement by 26 September 2024. In practice, this newly granted “grace period” will basically benefit serious laggards as compliant manufacturers had already transitioned to the EU MDR and obtained the corresponding Notified Body’s certificate for their QMS on or around 26 May 2021.
Custom-made devices are not CE marked. However, a manufacturer of custom-made devices must still follow EU MDR requirements. This includes implementation of Annex XIII, and registering in EUDAMED as an actor, even if UDI requirements do not apply. MDCG 2021-3 provides details on the different aspects of the EU MDR that a manufacturer of custom-made devices must observe.
What is a “significant change” under the EU MDR or IVDR?
A device loses its legacy status if it undergoes a significant change to design or intended purpose. If the device undergoes a significant change it must go through the EU MDR or IVDR approval process fulfilling all the relevant EU MDR or IVDR requirements, as applicable.
Design changes may sound simple and narrow, but the term is used in its broadest sense as can be seen below in the list summarizing changes identified in guidance documents MDCG 2020-3 (concerning the EU MDR) and MDCG 2022-6 (concerning the IVDR) on when change significance needs to be evaluated:
- Changes related to a corrective action, except if assessed and accepted by the competent authority where the manufacturer or its EC-REP resides.
- Changes to the device intended purpose, like extensions, new user/patient population, new way of clinical application.
- Changes in device design that alter the built-in control mechanism, the device’s operating principle, the source of energy or alarm systems, as well as any changes that may adversely affect the safety or performance and negatively impact the risk/benefit ratio.
- Most software changes.
- Certain changes in materials or medical device substances or IVD ingredients.
- Changes in terminal sterilization method or packaging design with impact on the sterile condition of the device.
For more details on significant changes under the EU MDR, see our blog article Medical device “significant changes” under the EU MDR & IVDR.
A device loses its legacy status if it undergoes a significant change to design or intended purpose.
What transitional periods and conditions apply to legacy devices?
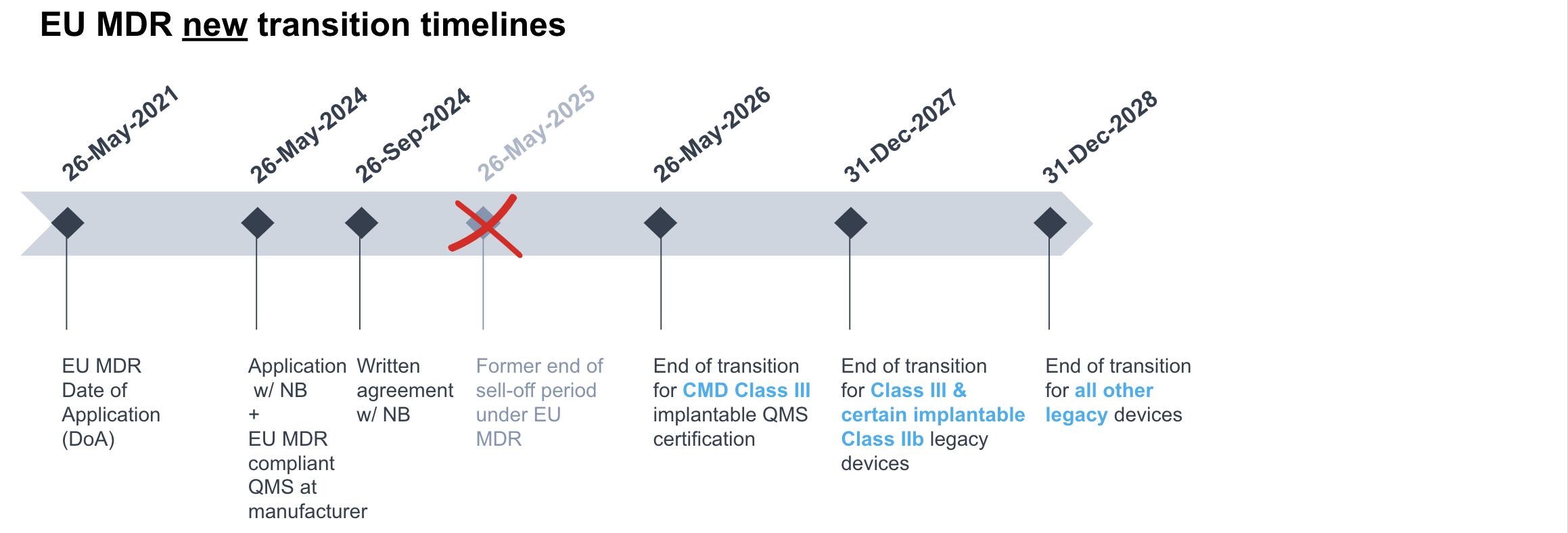
The below transitional timelines to upgrade CE-marking from the former Directives to the EU MDR or IVDR, as applicable, have been granted to legacy devices:
For non-IVDs with a valid certificate:
- 31 December 2027 for Class III devices and implantable Class IIb devices (except sutures, staples, dental fillings, dental braces, tooth crowns, screws, wedges, plates, wires, pins, clips and connectors).
- 31 December 2028 for other Class IIb devices, Class IIa devices, and Class Is/m devices.
For the purpose of these deadlines, the (future) classification, per EU MDR Annex VIII, applies.
Conditions, as detailed in EU MDR Article 120 (3c), include:
- the device continues to comply with Dir. 90/385/EEC or Dir. 93/42/EEC, whichever applies,
- no unacceptable risk for health and safety,
- no significant changes to the device design and intended purpose,
- manufacturer’s QMS in accordance with EU MDR Article 10(9) is in place no later than 26 May 2024,
- proof of formal application with a Notified Body for the transition to the EU MDR by 26 May 2024 and the written agreement with the Notified Body is signed no later than 26 September 2024.
For non-IVDs that did not require the involvement of a Notified Body under the MDD (e.g. Class I, EU MDR Annex XVI products) and would require so under the EU MDR: until 31 December 2028, per EU MDR Article 120 (3b), and subject to the same conditions listed above.
Per EU MDR Article 120(2), the same transitional periods as for non-IVDs with a valid certificate have now been granted for non-IVDs with an expired certificate before 20 March 2023, provided one of the below conditions are fulfilled:
- a written agreement had already been signed with the Notified Body for the EU MDR transition prior to the expiration of the certificate, or
- a competent authority has granted a derogation under EU MDR Article 59(1), or
- a competent authority has required the manufacturer to proceed with the applicable EU MDR conformity assessment procedure, per EU MDR Article 97(1).
See below for more details about expired certificates.
As already mentioned, implantable Class III custom-made devices, that under the EU MDR require a QMS certificate issued by a Notified Body, pursuant to EU MDR Article 120 (3f), they can remain on the market without such certificate until 26 May 2026, provided a formal application with a Notified Body has been lodged by 26 May 2024 and the written agreement with the Notified Body is signed no later than 26 September 2024.
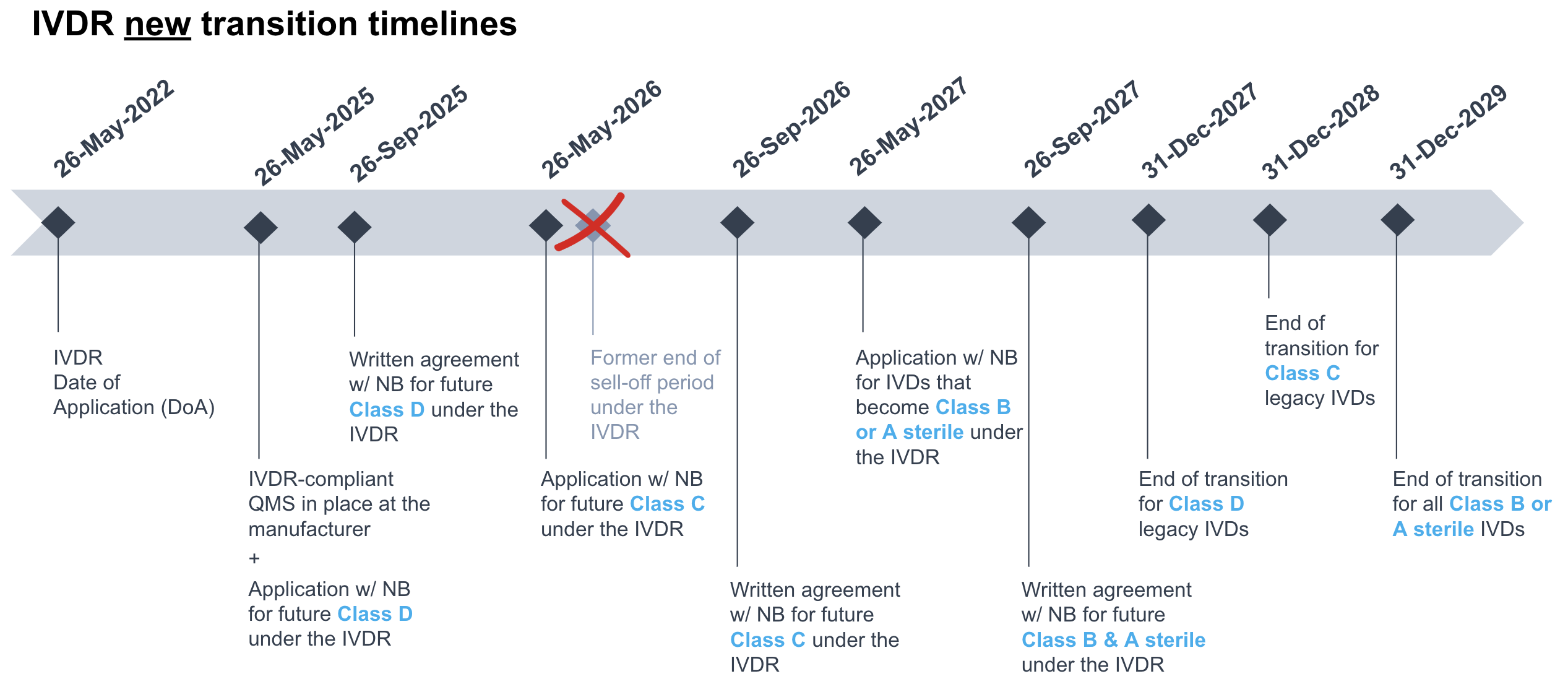
For IVDs with a valid certificate:
- 31 December 2027 for Class D IVDs.
- 31 December 2028 for Class C IVDs.
- 31 December 2029 for Class B and Class A sterile IVDs.
- For the purpose of these deadlines, the (future) classification, per IVDR Annex VIII, applies.
Conditions, as detailed in IVDR Article 110 (3c), include:
- the device continues to comply with Dir. 98/79/EC,
- no unacceptable risk for health and safety,
- no significant changes to the device design and intended purpose,
- manufacturer’s QMS in accordance with IVDR Article 10(8) is in place no later than 26 May 2025,
- proof of formal application with a Notified Body for the transition to the IVDR and written agreement with the Notified Body signed, within the following class-specific timelines:
- For Class D: application by 26 May 2025 and agreement by 26 September 2025.
- For Class C: application by 26 May 2026 and agreement by 26 September 2026
- For Class B and Class A sterile: application by 26 May 2027 and agreement by 26 September 2027.
In addition, as part of the amending Regulation (EU) 2023/607 to ease the bottleneck of the transition to the EU MDR and IVDR, the sell-off provisions in both the EU MDR and IVDR (i.e. basically one year after the end of each transitional period for upgrading CE-marking) are removed, so that stocks of legacy devices at distributors can be sold out indefinitely, beyond the end of the corresponding transitional period.
The below infographics summarize the new timelines under the EU MDR and IVDR:
Proof of compliance with the conditions to benefit of the transitional periods for legacy devices is to be demonstrated through:
- Manufacturer’s self-declaration confirming that the conditions for the extension are fulfilled, stating the end date of the transition period. A template for such self-declaration has been made available by MedTech Europe, and
- Notified Body’s confirmation letter stating the receipt of the manufacturer’s application for conformity assessment and the conclusion of a written agreement. Notified Bodies have no obligation to issue such letters, because the manufacturer could demonstrate the application for conformity assessment and concluded a written agreement with a notified body also by other means, such as a copy of the relevant documents.
What EU MDR or IVDR requirements apply to legacy devices?
EU MDR and IVDR provisions considered to apply to legacy devices are those detailed in the corresponding transitional provisions and are further detailed in guidance documents MDCG 2021-25 (concerning the EU MDR) and MDCG 2022-8 (concerning the IVDR), which include useful tables for the most relevant articles. In brief, duties that apply to legacy devices are those relative to post-market surveillance, market surveillance (i.e. deployed by the national competent authorities), vigilance, as well as registration of economic operators and devices.
All devices on the market are required to fulfill state-of-the-art requirements by securing compliance with all applicable standards and regulations.
Post-market surveillance and Vigilance
The most significant requirement for legacy devices is probably the post market surveillance. To be compliant with MDR/IVDR post market surveillance requires a systematic and proactive collection of data on the safety and performance of the device. EU MDR and IVDR also requires regular updates to the Periodic Safety Update Report.
|
Post-market surveillance plan (PMSP) |
MDR |
|
IVDR |
|
|
Post-market surveillance report (PMSR) |
MDR |
|
IVDR |
|
|
MDR |
|
|
IVDR |
|
|
Post-market Follow-up Plan (PMCFP) |
MDR |
|
IVDR |
|
|
Post-market Follow-up Report (PMCFR) |
MDR |
|
IVDR |
|
|
Vigilance reporting |
Per EU MDR Article 87 or IVDR Article 82:
Trend reporting, per EU MDR Article 88 or IVDR Article 83. |
* However, there is no requirement for a full revision of the technical documentation in accordance with Annexes II and III.
Note that for “old devices”, as described under When does a CE-marked device (medical device or IVD) qualify as legacy device? above, serious incident reporting under the EU MDR or IVDR applies. Such reporting would entail a specific EUDAMED registration of the old device manufacturer, as detailed in question # 5 of guidance document MDCG 2021-13.
Registration of economic operators and devices
In principle, the EU Commission is not in a position to make EUDAMED registration of economic operators and devices mandatory until EUDAMED is fully functional. This is now changing with the proposal to make EUDAMED mandatory in a gradual roll-out, which shall start by end of 2025.
Legacy devices must also be registered in EUDAMED (see MDCG 2019-5). If these devices do not have a Basic UDI-DI or an UDI-DI they must have EUDAMED DI (instead of the Basic UDI-DI) and EUDAMED ID (instead of the UDI-DI). If these devices are then transitioned to EU MDR or IVDR CE-marking, linkages should be created between the respective registrations.
Related obligations of economic operators
EU MDR or IVDR requirements that are not related to post-market surveillance, market surveillance, vigilance, registration of economic operators and devices should in principle not apply to economic operators in respect to legacy devices. Within this context, guidance documents MDCG 2021-25 (concerning the EU MDR) and MDCG 2022-8 (concerning the IVDR) consider that related provisions in chapters covering the duties of economic operators should be considered to apply, namely:
- for manufacturers under the EU MDR: Article 10(10) and Article 10(12) through 10(15);
- for manufacturers under IVDR: Article 10(9) and Article 10(11) through 10(14);
- for authorised representatives: Article 11(3)(c) through (g);
- for importers: Article 13(2) 2nd subparagraph, Article 13(4), Article 13(6) through 13(8), and Article 13(10);
- for distributors: Article 14(2) last subparagraph, and Article 14(4) through (6).

To be compliant with MDR/IVDR post market surveillance requires a systematic and proactive collection of data on the safety and performance of the device.
Do legacy device manufacturers need a PRRC?
The Person Responsible for Regulatory Compliance (PRRC) is a requirement introduced in Article 15 of the EU MDR and IVDR. If you want to learn more about the PRRC role, please read The role of a PRRC under the MDR, which can also be extrapolated to the IVDR.
Article 15 is not listed as part of EU MDR and IVDR transitional provisions applying to legacy devices. This is further confirmed in guidance documents MDCG 2021-25 and MDCG 2022-8, where Article 15 is cited as an example of provisions that do not apply to legacy devices.
When are Procedure Packs and Systems considered “legacy”?
Systems and procedure packs are considered legacy devices when they consist only of legacy devices and if a Statement per MDD Article 12 has been drawn up for them prior to 26 May 2021.
Once a component of the system or procedure pack transitions to CE-marking under the EU MDR or IVDR, the entire system or procedure pack loses its legacy status. However, any legacy devices included within a EU MDR system or procedure pack are covered by the transitional provisions of EU MDR Article 120(3) or IVDR Article 110(3), as applicable.
What specifically needs to be considered for legacy devices in Switzerland?
Switzerland is considered a third country to the effects of the EU MDR since 26 May 2021 and of the IVDR since 26 May 2022. Swiss legislation was adapted accordingly but it recognizes CE-marking per the EU MDR under the Swiss Medical Device Ordinance (MedDO, SR 812.213), and per the IVDR, under the Swiss In-vitro Diagnostic Ordinance (IvDO, SR 812.219).
As such, “legacy devices” and “old devices” have the same meaning and implications as under the EU MDR and IVDR. Because Swiss legislation basically points out the corresponding provisions in the EU MDR and IVDR, relevant updates in the Regulations result in subsequent reviews of the MedDO and IvDO, respectively.
“Legacy devices from manufacturers based outside Switzerland need a Swiss Authorised Representative”
In a nutshell, the requirements for legacy devices are the same in Switzerland as in the EU. However, because Switzerland is now a third country, all legacy devices from manufactures based outside Switzerland need a Swiss Authorised Representative, which is a new economic operator role instated since 26 May 2021 for non-IVDs and 26 May 2022 for IVDs. In addition, any Swiss entity bringing devices from abroad into Switzerland and further supplying them within the country becomes de facto Swiss importer and endorses new duties under the MedDO and IvDO. These requirements apply regardless of the legacy status or transitional stage of the device.
Moreover, Swissmedic expects Swiss Authorised Representatives and Swiss importers to assess the plausibility of extended certificate validity of legacy devices by verifying the manufacturer’s self-declarations and the Notified Body’s confirmation letters. For more details, see question (d) on page 18 of Swissmedic’s Info Sheet on the Obligations of Economic Operators.
How Decomplix can help
Decomplix offers consultancy services in all regulatory and quality management matters relative to medical devices, with particular focus on the Swiss market and CE marking.
We can support your company in fulfilling all the requirements of under the EU MDR or IVDR and ensure compliance in the most efficient manner. If you wish to discuss your needs with us, please contact us for a non-binding CE marking quote.
In addition, we offer Swiss Authorised Representative services under both the MedDO and the IvDO. Our services include instructions and checklists for your understanding of the applicable requirements. If you are interested in Decomplix’ Swiss Authorised Representative services, please contact us via this form and send us the relevant data mentioned there.
Further reading
- Is my product a medical device in Europe? How to determine if your product requires medical device CE marking
- How to obtain a CE mark for a medical device
- Class I medical device requirements for manufacturers under the EU MDR
- Guidance on medical device significant changes
- Swiss authorised representatives for medical device manufacturers



