
Swiss authorised representatives for medical device manufacturers
Have you heard about Swiss Authorised Representatives for medical device manufacturers yet? Here is all you need to know about it.
Replaces version of 28.04.2023 The agreement facilitating trade of CE-marked products between the European Union (EU) and Switzerland, known as the Mutual Recognition Agreement (MRA), ceased to apply with the application of the new Regulation (EU) 2017/745 on medical devices (EU MDR), andthe new Regulation (EU) 2017/746 on IVDs (IVDR). As a result, Switzerland is now considered a third country to the effects of the EU MDR and IVDR. In parallel, Switzerland enacted a revised version of its Medical Device Ordinance, SR 812.213 (MedDO) and issued a new In-Vitro Diagnostics Ordinance, SR 812.219 (IvDO), adapted to the new situation. Amongst the new requirements introduced by the MedDO and the IvDO, manufacturers who are not based in Switzerland, which now includes those based in EEA countries (except Liechtenstein) and those having a EU Authorised Representative in a EEA country, require a Swiss Authorised Representative (in German: Schweizer Bevollmächtigter, and in French: mandataire suisse) to continue placing their CE-marked medical devices on the market in Switzerland. Since all “grace periods” to appoint a Swiss Authorised Representative that had been granted in the MedDO and IvDO have now expired, medical devices (incl. IVDs) can no longer be placed on the Swiss market if their manufacturer does not have a Swiss Authorised Representative. This applies equally to “legacy devices”. If you are not familiar with the role of and requirements for appointing a Swiss Authorised Representative for foreign medical device manufacturers, read on. You can also find answers to frequently asked questions that we are currently receiving from various customers as Decomplix provides Swiss Authorised Representative services.
Key Takeaways
- The rules for placing medical devices or IVDs on the Swiss market have significantly changed since 26 May 2021 for medical devices that are not IVDs and since 26 May 2022 for IVDs.
- Any foreign manufacturer that places medical devices and IVDs on the Swiss market must officially appoint a Swiss Authorised Representative and, although “grace periods” were granted, these have now expired.
Content
What has happened with medical devices in Switzerland? What is the MRA? What are the MedDO and the IvDO?
Similar to the situation in the United Kingdom after BREXIT, the requirements for non-Swiss manufacturers of CE-marked medical devices got a little more complicated after 26 May 2021, and for in-vitro diagnostic devices (IVDs), after 26 May 2022. Importing CE-marked medical devices (incl. IVDs) into Switzerland used to be indistinguishable from importing them into any other EEA country, because the former European Directives 93/42/EEC concerning medical devices (MDD), 90/385/EEC on active implantable medical devices (AIMDD), and 98/79/EC on in vitro diagnostic medical devices (IVDD) were fully adopted under the former Swiss Medical Device Ordinance (oMedDO, SR 812.213), based on the Mutual Recognition Agreement (MRA) between Switzerland and the EU. This is no longer the case. As communicated in the Notice to Stakeholders issued by the EU Commission, the MRA ceased to apply for medical devices on 26 May 2021, date of application of the EU MDR. Since then, Switzerland is considered a third country to the effects of the EU MDR. On the same date, Switzerland enacted a revised version of its Medical Device Ordinance, SR 812.213 (MedDO), which introduced additional requirements to account for the absence of an updated MRA. Per Article 51 of this revised MedDO, medical device manufacturers established in EEA countries (except Liechtenstein), or established elsewhere and represented by EU Authorised Representative located in a EEA country, now need a Swiss Authorised Representative to sell their CE-marked devices in Switzerland, just like medical device manufacturers from outside the Union market. The same requirement was introduced for in vitro diagnostic medical devices (IVDs) under Article 44 of the new Swiss In-vitro Diagnostic Medical Device Ordinance, SR 812.219 (IvDO) that came into effect on 26-May-2022, simultaneously to the date of application of the IVDR.

The appointment of an authorised representative cannot happen overnight.
Furthermore, because the new EU MDR repealed the former MDD and AIMDD and the IVDR repealed the former IVDD, the existing MRA covering medical devices and IVDs is obsolete, and a Swiss Authorised Representative is required both for devices CE marked under the new EU MDR and IVDR and for so-called “legacy devices”. Legacy devices are those that had been CE marked under the previous Directives (MDD, AIMDD or IVDD) and benefit from the transitional provisions in Article 120 of the EU MDR or Article 110 of the IVDR, as applicable. Read more about “Legacy” medical devices and IVDs under EU legislation. In order to revert the situation, the MRA would need to be updated, which is unlikely to happen anytime soon as the EU Commission will not negotiate or update the MRA until an agreement between both parties has been established. Switzerland’s decision to not sign and to stop the process of the institutional agreement (InstA) unilaterally has wide-ranging and long-term consequences. The current political situation in Switzerland makes it thus highly unlikely that there will be a solution in the foreseeable future. Without an updated MRA, Switzerland will remain as a “third country” to the effects of the EU MDR and IVDR, and EEA countries will remain as “third countries” under the Swiss legislation on medical devices and IVDs.
What is a Swiss Authorised Representative?
Per the definition in Article 4 §1(g) of the Swiss MedDO and Article 4 §1(f) of the Swiss IvDO, a Swiss Authorised Representative for a foreign medical device manufacturer corresponds to: “any natural or legal person established within Switzerland who has received a written mandate from a manufacturer located in another country to act on the manufacturer’s behalf in relation to specified tasks with regard to the latter’s obligation under this Ordinance”. Upon acceptance of the written mandate, this natural or legal person based in Switzerland becomes the representative of the foreign manufacturer before the Swiss competent authority, Swissmedic, and is legally liable for defective devices, on the same basis and jointly and severally with the manufacturer. Swissmedic has clarified that, by virtue of the customs treaty with Liechtenstein, “established in Switzerland” means both Switzerland and Liechtenstein, provided the devices are placed on the market based on the MedDO/IvDO. Moreover, per Article 51 §2 of the MedDO and Article 44 §2 of the IvDO, the Swiss Authorised Representative is responsible for the formal and safety-related aspects of placing a device on the Swiss market.
Is a Swiss Authorised Representative needed for all types of medical devices?
A Swiss Authorised Representative is required under the MedDO for:
- all medical devices, including custom-made devices and products without a medical purpose listed in Annex 1 of the MedDO,
- all Procedure Packs, as defined in Article 2(10) of the EU MDR, and
- all Systems, as defined in Article 2(11) of the EU MDR
that are being placed on the market in Switzerland, within the meaning of Article 4 §1(b) of the MedDO. In addition, a Swiss Authorised Representative is required for all in vitro diagnostic devices (IVDs) placed on the market in Switzerland, within the meaning of Article 4 §1(b) of the new IvDO. In theory, direct sales of medical devices and IVDs from foreign manufacturers to Swiss-based healthcare institutions who are the end users of the devices could be possible without designation of a Swiss Authorised Representative because this direct sale does not correspond to the definition of “placing on the market”. However, Swissmedic discourages such practices. Chapter 7 of Swissmedic’s Information Sheet on Procurement of Medical Devices in Health Institutions indicates that “healthcare professionals and institutions should usually procure products from a Swiss manufacturer or that have a Swiss authorised representative and only directly use products from abroad without a Swiss authorised representative in justified exceptional cases”. Note that the requirement to appoint a Swiss Authorised Representative applies equally to devices placed on the market in compliance with the new Regulations (i.e. EU MDR or IVDR) and to “legacy devices” that may remain on the market in compliance with the former MDD/AIMDD/IVDD, including Procedure Packs or Systems still covered under the MDD.
What are the duties of the Swiss Authorised Representative?
The duties of a Swiss Authorised Representative under the MedDO/IvDO are similar to those of a EU Authorised Representative under the EU MDR/IVDR. Article 51 §3 of the MedDO describing the Swiss Authorised Representative’s duties calls out Article 11 of the EU MDR and Article 44 §3 of the IvDO calls out Article 11 of the IVDR. Consequently, the Swiss Authorised Representative role for medical devices is analogous to that of the EU Authorised Representative. Similarly, per Article 51 §4 of the MedDO and Article 44 §5 of the IvDO, a change in Swiss Authorised Representative is governed by the same requirements as those for a change in EU Authorised Representative, per Article 12 of the EU MDR or IVDR, as applicable. It is however important to highlight a significant difference between the role of EU Authorised Representative under the EU MDR and that of Swiss Authorised Representative under the MedDO: as previously indicated, according to Article 51 §2 of the MedDO and Article 44 §2 of the IvDO, the Swiss Authorised Representative is “responsible for the formal and safety-related aspects of placing the device on the market”. Although there is no guidance that allows clarifying this addition with respect to the EU MDR/IVDR, the level of responsibility of the Swiss Authorised Representative appears to be higher than that of its EU counterpart. Pursuant to Article 51 §3 of the MedDO (respectively Article 11 of the EU MDR) and Article 44 §3 of the IvDO (respectively, Article 11 of the IVDR), the duties of a Swiss Authorised Representative include:
- To verify that the Declaration of Conformity and technical documentation have been drawn up and, where applicable, that the manufacturer has carried out an appropriate conformity assessment procedure. This would include verifying that the device labeling fulfills the requirements of the MedDO/IvDO. In addition, for the verification of extended validity of EC Certificates for “legacy” devices under the MDD/AIMDD. Swissmedic’s expectation is for Swiss Authorised Representatives to check the plausibility of:
- the manufacturer’s self-declaration under EU MDR Art. 120(3c),
- the Notified Body’s confirmation letter stating that the manufacturer has lodged an application for EU MDR certification.
- To keep available a copy of the technical documentation, the Declaration of Conformity and, if applicable, a copy of the relevant certificate, including any amendments and supplements, at the disposal of the competent authority (Swissmedic) for the applicable retention period (10 or 15 years, depending on the type of device). As an alternative, per Article 51 §3bis of the MedDO and Article 44 §4 of the IvDO, instead of the Swiss Authorised Representative keeping a copy of the technical documentation, the manufacturer could submit the documentation directly to Swissmedic on request, in which case the Swiss Authorised Representative shall ensure that this happens within 7 days of Swissmedic’s request.
- To comply with the applicable Economic Operator registration obligations, which in Switzerland are provided for in Article 55 of the MedDO and Article 48 of the IvDO, whichever applies.
- Upon request from Swissmedic, to provide all the information and documentation necessary to demonstrate the conformity of a device, which might be required in the Swiss official languages (i.e. German, French, and Italian).
- To forward to the manufacturer any request from Swissmedic for either device samples or access to a device, and to verify that Swissmedic receives those samples or is given access to the device.
- To cooperate with Swissmedic on any preventive or corrective action taken to eliminate or, if that is not possible, mitigate the risks posed by the medical devices.
- To immediately inform the manufacturer about complaints and reports from healthcare professionals, patients and users about suspected incidents related to a device for which they have been designated.
- To terminate the mandate if the manufacturer acts contrary to its obligations under the MedDO.
In addition, the Swiss Authorised Representative is responsible for ensuring the reporting to Swissmedic any:
- Serious incidents occurring in Switzerland, as soon as it becomes aware of them, as well as the corresponding final report with any investigation findings and indication of corrective actions taken,
- Field Safety Corrective Actions (FSCA) initiated in Switzerland, and
- Vigilance trend reports on incidents in Switzerland and abroad.
This responsibility, which applies under the MedDO and IvDO alike, appears to be understood by Swissmedic as an oversight role, where the actual reporting can be undertaken by either the foreign manufacturer or the Swiss Authorised Representative. However, per Article 66 §2bis of the MedDO and Article 59 §3 of the IvDO, the Swiss Authorised Representative is expected to submit any final reports and the trend reports without being requested to do so, and such responsibility transfer from the manufacturer to the Swiss Authorised Representative must be agreed in writing in the mandate, which requires some means of control of the Swiss Authorised Representative over the manufacturer’s Vigilance process. For more details on Vigilance requirements, please consult Swissmedic’s Vigilance webpage.

The level of responsibility of the Swiss Authorised Representative appears to be higher than that of its EU counterpart.
Also, per Article 55 §1 of the MedDO and Article 48 §1 of the IvDO, the Swiss Authorised Representative needs to register for its economic operator role with Swissmedic and its initial registration must be completed at the latest 3 months after the first placing of devices on the Swiss market (including Liechtenstein), which involves obtaining a Swiss Registration Number (CHRN) from Swissmedic. Any subsequent changes to the Swiss Authorised Representative’s information notified to Swissmedic (e.g. change in address, change in PRRC) shall be submitted by the Swiss Authorised Representative within 1 week. There are certain manufacturer’s obligations that could not be delegated to the Swiss Authorised Representative either under the MedDO or the IvDO, namely:
- Ensuring the device conformity.
- Implementing a compliant risk management system.
- Performing clinical evaluations, including post-market clinical follow-up activities.
- Compiling or keeping up-to-date the device technical documentation.
- Drawing up the EU Declaration of Conformity.
- Assigning and maintaining device UDIs.
- Maintaining the manufacturer’s quality management system.
- Implementing the manufacturer’s post-market monitoring system.
- Preparing device labeling (i.e. on-device labels, packaging labels, and instructions for use, as applicable).
- Establishing any necessary corrective actions to bring any non-conforming device into conformity.
Because both the MedDO and the IvDO require, per their respective Article 6, compliance with the General Safety and Performance Requirements (GSPR) in Annex I of the EU MDR or IVDR, which in turn require that the particulars of the EU Authorised Representative be indicated on the device label, the Swiss Authorised Representative’s name and address would need to appear on the device labelling. However, exceptions and “grace periods” apply. For details, see: Do foreign manufacturers need to change the device labelling? Also for “legacy devices”? The Swiss Authorised Representative must register with Swissmedic in order to obtain a Swiss Registration Number (CHRN). for its economic operator role. This registration, required per Article 55 of the MedDO and Article 48 of the IvDO must be completed within 3 months of the first placing devices on the Swiss market (or in Liechtenstein). Subsequent changes to the Swiss Authorised Representative’s information (e.g. change in address) need to be notified to Swissmedic within 1 week. Last, as required under Article 52 of the MedDO and Article 45 of the IvDO, the Swiss Authorised Representative must have permanently and continuously at its disposal a Person Responsible for Regulatory Compliance. This role mirrors the Person Responsible for Regulatory Compliance (PRRC) under Article 15 of the EU MDR/IVDR. However, contrary to the position expressed in guidance document MDCG 2019-7 relative to the PRRC role in EU, Swissmedic does not limit the geographic location of the Swiss Authorised Representative’s PRRC. As such, the Swiss Authorised Representative’s PRRC can be based in any country and can even correspond to the PRRC of the foreign manufacturer or of its EU Authorized Representative. Read more about the role of a PRRC under the EU MDR & IVDR.
What UDI registration requirements apply to the Swiss Authorised Representative under the MedDO?
EUDAMED not being accessible to Swissmedic as the competent authority of a “third country” for the effects of the EU MDR and IVDR, the registration of UDI core data elements required under Article 29 of the EU MDR or Article 26 of the IVDR will be implemented in Switzerland via the country-specific medical device information system (swissdamed), according to Article 62c of the Swiss Therapeutic Product Act (TPA, SR 812.21). This database will encompass 2 modules (economic operator registration and device registration) and is still under development. The details of such registration will be governed by the MedDO/IvDO, which will have to be amended accordingly. Although swissdamed is expected to go live in 2024, at this stage, it is not known exactly when and how it will be implemented. For the time being, the only devices from foreign manufacturers that need to be notified to Swissmedic are Custom-made devices (CMD), per MedDO Art. 19. According to Swissmedic’s FAQs on the notification of medical devices, the obligation to notify CMDs applies to either Swiss Authorised Representative’s, Swiss importers or Swiss distributors before making CMDs available on the Swiss market. This requirement applies also to Swiss Authorised Representatives, importers or distributors based in Liechtenstein, when the devices are placed on the market according to the MedDO. If you are interested in Decomplix CMD notification services, please contact us via this form.
Is the Swiss Authorised Representative also the Swiss Importer?
No, the Swiss Authorised Representative and the Swiss Importer are different economic operator roles with different regulatory duties under the MedDO and IvDO. The duties of the Swiss Importer for medical devices are governed by Article 53 of the MedDO and Article 46 of the IvDO. Even if these involve compliance verification and notification requirements, which could be considered akin to those applicable to the Swiss Authorised Representative, the obligations cover also warehousing and transportation activities, including the necessary quality management system. Whereas a Swiss Importer could take on the additional role of Swiss Authorised Representative, provided it had sufficient in-house regulatory resources, the opposite is rarely the case since importation entails, in addition to regulatory compliance with the MedDO/IvDO, customs clearance, logistic, and tax-related activities and companies offering Swiss Authorised Representative services tend to be regulatory consultancy companies without competence in those domains. The role of a Swiss Importer is naturally easier for an existing Swiss distributor. It is also important to note that Swissmedic, in its Information Sheet on the Obligation of Economic Operators, considers an entity to be the Swiss Importer “by action” (i.e. any entity who places a medical device on the Swiss market) as opposed to “by designation”. The duties of a Swiss Importer under the MedDO/IvDO are similar to those of the EU importer under the EU MDR/IVDR. Like the Swiss Authorised Representative, the Swiss Importer shall register with Swissmedic within 3 months of its placing of devices (MDR/IVDR compliant or “legacy” devices) on the Swiss market for the first time after the date of implementation of the MedDO/IvDO.

The role of a Swiss Importer is naturally easier for an existing Swiss distributor.
Do foreign manufacturers need to change the device labelling? Also for “legacy devices”?
The MedDO and IvDO do not explicitly require that the Swiss Authorised Representative’s particulars appear on the device labelling. Now, Article 6 §2 of the MedDO and of the IvDO requires that all medical devices placed on the Swiss market comply with the General Safety & Performance Requirements (GSPR) in Annex I of the EU MDR/IVDR, which in turn require that the product label bears the name and address of the EU Authorised Representative. This applies by analogy, specifically because MedDO Annex 2 and IvDO Annex 1 list the correspondence of terms between the EU regulation and Swiss ordinance. Amongst them, “Union” would correspond to “Switzerland”. Therefore, GSPR #23.2(d) in the EU MDR and GSPR #20.2(d) in the IVDR should be “translated” to the effects of MedDO/IvDO into: “if the manufacturer has its registered place of business outside Switzerland, the name of the authorised representative and address of the registered place of business of the authorised representative.“ As to “legacy devices”, per Article 101 of the MedDO and Article 82 of the IvDO, they can remain on the Swiss market, provided they continue to comply with the requirements of the MDD/AIMDD or IVDD, as applicable. And the Essential Requirements (ER) in Annex I of the MDD/IVDD or Annex 1 of the AIMDD also call for the authorised representative’s name and address to be mentioned on the device labelling. The indication of the Swiss Authorised Representative’s particulars is required from the day of application of the MedDO/IvDO but some exceptions or grace periods were granted by Swissmedic. The currently applicable requirements are summarized in its Information Sheet on the Obligations of Economic Operators, as follows:
Type of device |
Labelling requirement |
| EU MDR compliant devices (all classes) | On the label |
| MDD/AIMDD compliant devices that have a EEA-based legal manufacturer or EC-REP | MDD: On the label or in the instructions for use or on a document accompanying the device. (*)AIMDD: On the commercial packaging and in the instructions for use or on a document accompanying the device. (*) |
| MDD/AIMDD compliant devices that do not have a EEA-based legal manufacturer or EC-REP | MDD: On the label or instructions for use.AIMDD: On the sales packaging and instructions for use. |
| IVDR compliant IVDs NOT intended for self-testing | Until 31-Mar-2025: Either on the label or on a document accompanying the device.(*)After 31-Mar-2025: On the label. |
| IVDR compliant IVDs intended for self-testing | On the label |
| IVDD compliant devices that have a EEA-based legal manufacturer or EC-REP | On the labelling, on the external packaging, on the instructions for use, or on a document accompanying the device. (*) |
| IVDD compliant devices that do NOT have a EEA-based legal manufacturer or EC-REP | On the labelling, on the outer packaging, or instructions for use. |
(*) Swissmedic considers that the “document accompanying the device” may be attached to or separate from the device. It does not therefore necessarily have to reach the end user. The purpose of the information is a quick and clear identification of the economic operators responsible for the devices at hand, e.g. for the implementation of product recalls, for the reporting of incidents, for notifications of dangerous products or non-conformities and in the context of enforcement. Examples of documents accompanying the device: delivery note, warranty certificate, customs documents, invoice, a sticker on the packaging or instructions for use. Note that the Swiss Authorised Representative’s labelling requirement applies to any device unit placed on the Swiss market, not for a device model. As such, it is required for new devices as well as for refurbished devices that are assigned a new serial number or UDI-PI. In the cases where the Swiss Authorised Representative’s particulars must appear on the device labelling, Decomplix considers that a permanent sticker with the Swiss Authorised Representative’s particulars would suffice, and the sticker should be affixed to the existing packaging element that requires such labelling. The over-labelling could be undertaken by the Swiss importer. When required on the labelling, the Swiss Authorised Representative’s particulars must be preceded by either the new symbol below or any of the following texts: “CH authorised representative” / “Authorised representative for Switzerland” / “CH-REP”.
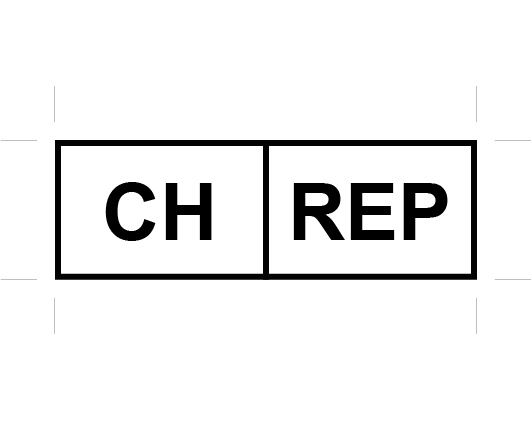
The new Swiss Authorised Representative (CH-REP) symbol.
According to Article 16 §2 of the MedDO and Article 15 §2 of the IvDO relative to language requirements that apply to labelling, the first two texts would require translation in German, French and Italian. Conversely, the use of the “CH-REP” symbol or text would not. As to the actual address, according to Swissmedic’s explanation on its CH-REP webpage, the main requirement is that the address “must enable contact to be established”. Decomplix considers that it is not necessary to translate the city in the Swiss Authorised Representative’s address into other Swiss languages as addresses within Switzerland are driven by the postal code. Also, from Swissmedic’s perspective, it is acceptable not to explain the meaning of this Swiss-specific CH-REP symbol in the instructions for use.
What happens with Procedure Packs and Systems?
Assemblers of medical device kits that correspond to the definition of Procedure Packs, per Article 2(10) of the EU MDR, or Systems, per Article 2(11) of the EU MDR, also need to appoint a Swiss Authorised Representative, per Article 51 §5 of the MedDO. This might come as a surprise to non-EEA Procedure Pack or System producers because there is no similar requirement at the EU level. The MedDO does not provide any further details on the implications. Neither has Swissmedic provided official guidance on its expectations yet. For example:
- What exactly should the Swiss Authorised Representative verify? Should the verification be limited to the statement under Article 22 of the EU MDR and the label? Should it include the verification of proof of mutual compatibility and/or verification of the conformity of the individual products contained in the Procedure Pack or System?
- Is the Swiss Authorised Representative of a Procedure Pack or System expected to become also the Swiss Authorised Representative of the individual CE-marked devices therein? Unofficial information from Swissmedic appears to indicate that this is not the case. Then, is the Swiss Authorised Representative of a Procedure Pack or System expected to verify that a Swiss Authorised Representative exists already for each individual CE-marked device therein?
- Is the Swiss Authorised Representative of a Procedure Pack or System expected to coordinate Vigilance reporting with the Swiss Authorised Representatives of the individual CE-marked devices therein?
In the absence of clarification there is a significant risk of uneven interpretation of the requirements and chaotic implementation across the entities offering Swiss Authorised Representative’s services. For this reason, Decomplix currently exerts extreme caution in endorsing the Swiss Authorised Representative role for Procedure Pack or System assemblers who are not also the legal manufacturer of the individual CE-marked devices therein, until Swissmedic brings clarity to this topic.

Assemblers of medical device kits that correspond to the definition of Procedure Packs or Systems also need to appoint a Swiss Authorised Representative
How is other EU legislation that might be relevant for medical devices affected by the MRA?
Similar to the situation of medical devices and IVDs, it is safe to surmise that as long as the EU legal act in the scope of Annex 1 of the MRA does not change, the mutual recognition remains valid because the MRA has not been withdrawn. Annex 1 of the MRA covers the following product sectors:

On 26 May 2022, Directive 98/79/EC on in-vitro diagnostic devices (IVDD) which is covered in the MRA was replaced by the new Regulation 2017/746 (IVDR)
Out of these, some might apply to medical devices beyond Chapter 4, e.g. machinery, personal protective equipment, radio equipment, equipment for use in potentially explosive atmospheres, and it is important for foreign manufacturers to monitor any upcoming changes that might entail the repeal of the existing EU legislative acts. The next one to be affected might be the Machinery Directive (i.e. Dir. 2002/46/EC), as there is a EU Commission proposal for a new Regulation. There is no information for now as to how Switzerland will prepare for it.
What actions do foreign manufacturers need to take on the Swiss market?
The appointment of the Swiss Authorised Representative does not happen overnight. Even once the manufacturer has identified the most convenient entity in Switzerland, the designation process entails, at a minimum:
- Swiss Authorised Representative mandate review and, where necessary, negotiation of the related contractual clauses. This might be very time consuming, particularly for EEA-based manufacturers who are not familiar with the requirements of a EU Authorised Representative and for foreign manufacturers who have not read the MedDO/IvDO.
- Setting up internal procedures in the foreign manufacturer’s Quality Management System to describe, for example the communication processes, the Swiss-specific Vigilance requirements, the labelling requirements, and the availability of the Technical Documentation in English or a Swiss national language within 7 days of a Swissmedic’s request.
- Agreeing on the logistics to ensure the most suitable means of sharing information and documents.
- Undergoing the initial verification of compliance by the Swiss Authorised Representative, which might result in a rejection of the Swiss Authorised Representative mandate, which is a legal obligation under Article 11(3)(h) of both the EU MDR and IVDR. Such a rejection would imply that the foreign manufacturer would have to seek another Swiss Authorised Representative and start the process over, without guarantee of a more successful outcome.
- Implementing the Swiss Authorised Representative’s particulars on the device labelling, if and when required.
Note that a foreign manufacturer may appoint more than one Swiss Authorised Representative to cover its portfolio intended to be sold in Switzeland, which might make sense for big corporations with independent divisions. However, the designation of each Swiss Authorised Representative must be effective at least for all devices of the same generic device group sold in Switzerland.
How can Decomplix help?
Decomplix offers Swiss Authorised Representative services under both the MedDO and the IvDO, since May 2021, is duly registered with Swissmedic, and already represents numerous foreign manufacturers of different sizes all over the world. Our services include instructions and checklists for your understanding of the applicable requirements.
If you are interested in Decomplix’ Swiss Authorised Representative services, please contact us via this form and send us the relevant data mentioned there.



