
“Legacy”-Medizinprodukte und -IVD nach EU-Regulierung
Medizinprodukte und In-vitro-Diagnostika (IVD), die noch nach den alten EU-Richtlinien CE-gekennzeichnet sind, können unter gewissen Bedingungen der neuen EU-Verordnungen weiterhin in Verkehr gebracht werden. Solche Produkte werden als “Legacy-Produkte” bezeichnet.
Wenn Sie unsicher sind, ob Ihr Produkt als Legacy-Produkt gilt, oder wenn Sie mehr darüber wissen möchten, wie Legacy-Produkte im Rahmen der EU-MDR oder IVDR geregelt sind, lesen Sie weiter.
Ersetzt die Version vom April 2023
Wichtige Erkenntnisse:
- Wenn Ihr Produkt der Risikoklasse I oder Ihr selbst deklariertes IVD keine Benannte Stelle gemäss der EU-MDR oder IVDR erfordert, muss es bereits mit der neuen Verordnung konform sein (es gilt in diesem Fall nicht als Legacy-Produkt).
- Veränderungen am Produktdesign oder der Zweckbestimmung führen zum Verlust des “Legacy”-Status. Vergewissern Sie sich, dass Ihr Änderungsmanagement dies berücksichtigt.
- Die Übergangsfristen für Legacy-Produkte haben sich geändert. Informieren Sie sich in diesem Artikel über die neusten Fristen und geltenden Bedingungen. Und vergessen Sie nicht, dass ausländische Hersteller für alle Legacy-Produkte, die in der Schweiz verkauft werden, einen Schweizer Bevollmächtigten benötigen.
Inhalt:
Wann gilt ein CE-gekennzeichnetes Produkt (Medizinprodukt oder IVD) als Legacy-Produkt?
Legacy-Produkte im Sinne der EU-MDR und der IVDR sind Produkte, die nach dem Datum des Inkrafttretens der entsprechenden Verordnung (d.h. dem 26. Mai 2021 für die EU-MDR und dem 26. Mai 2022 für die IVDR) in Verkehr gebracht werden dürfen. Voraussetzung ist, dass die folgenden Punkte der Übergangsbestimmungen erfüllt sind (Art. 120 Abs. 3 EU-MDR bzw. Art. 110 Abs. 3 IVDR):
- das Medizinprodukt oder IVD verfügt noch über eine gültige Bescheinigung der Benannten Stelle (ausgestellt gemäss der früheren MDD, AIMDD oder IVDD),
- das Medizinprodukte (exklusive IVD) hat eine Bescheinigung der Benannten Stelle, die vor dem 20. März 2023 abgelaufen ist, und
- das Medizinprodukt oder IVD benötigte nach den früheren Richtlinien keine Benannte Stelle für die CE-Kennzeichnung (d.h. keine Bescheinigung), gemäss EU-MDR bzw. IVDR hingegen schon.
Dies bedeutet, dass die folgenden Kategorien NICHT als Legacy-Produkte gelten:
- Produkte der Klasse I nach der MDD, die auch nach der EU-MDR Produkte der Klasse I bleiben und keinen Einbezug einer Benannten Stelle erfordern. Letzteres ist bei Produkten der Fall, die weder eine Messfunktion haben, noch steril oder chirurgisch wiederverwendbar sind.
- Selbstdeklarierte (nicht sterile) IVD gemäss der IVDD, die IVD (nicht steril) der Klasse A gemäss der IVDR entsprechen.
Beachten Sie, dass Produkte, die vor dem Datum des Inkrafttretens der EU-MDR bzw. IVDR in Verkehr gebracht wurden (26. Mai 2021 bzw. 26. Mai 2022), als “Altprodukte” und nicht als “Legacy-Produkte” gelten. Obwohl Altprodukte auf dem Markt lange Zeit im Einsatz bleiben können (z.B. chirurgische Geräte oder Laborzentrifugen für IVD-Tests), unterliegen sie nicht den Anforderungen der EU-MDR bzw. IVDR, mit Ausnahme der Meldung schwerwiegender Vorkommnisse. Weitere Informationen finden Sie im Abschnitt Welche EU-MDR- oder IVDR-Anforderungen gelten für Legacy-Produkte?
Es gibt drei Situationen, in denen Legacy-Produkte ihren Legacy-Status verlieren können:
- Wenn wesentliche Änderungen am Produktdesign oder bei der Zweckbestimmung vorgenommen werden. Siehe Abschnitt Was ist eine “wesentliche Änderung” nach EU-MDR und IVDR?
- Wenn die entsprechenden Übergangsfristen in der EU-MDR oder IVDR ablaufen. Siehe Abschnitt Welche Übergangsfristen und -bedingungen gelten für Legacy-Produkte?
- Wenn die nach den früheren Richtlinien ausgestellte Bescheinigung der Benannten Stelle abläuft, ohne dass die Bedingungen für die Inanspruchnahme der Übergangsfristen in der EU-MDR oder IVDR erfüllt wurden. Siehe Abschnitt Welche Übergangsfristen und -bedingungen gelten für Legacy-Produkte?
Wie sieht es mit Sonderanfertigungen aus?
Die EU-MDR (Art. 2 Abs. 3) definiert eine Sonderanfertigung als:
“ein Produkt, das speziell gemäss einer schriftlichen Verordnung einer aufgrund ihrer beruflichen Qualifikation nach den nationalen Rechtsvorschriften zur Ausstellung von Verordnungen berechtigten Person angefertigt wird, die eigenverantwortlich die genaue Auslegung und die Merkmale des Produkts festlegt, das nur für einen einzigen Patienten bestimmt ist, um ausschliesslich dessen individuelle Zustand und dessen individuellen Bedürfnissen zu entsprechen.”
Bei Sonderanfertigungen handelt es sich also um Medizinprodukte, deren Produktdesign auf die Bedürfnisse eines bestimmten Patienten zugeschnitten ist (d.h.: keine Massenproduktion). Ein klassisches Beispiel für eine Sonderanfertigung ist eine Gliedmassenprothese, die auf Verordnung einer autorisierten Person (z.B. eines Orthopäden) hergestellt wird.
«Hersteller von Sonderanfertigungen müssen die Anforderungen der EU-MDR einhalten, auch wenn keine Zertifizierung erforderlich ist»
Sonderanfertigungen fielen nicht unter die ursprünglichen Übergangsbestimmungen in Art. 120 der EU-MDR. Daher galten sie nicht als Legacy-Medizinprodukte und mussten ab dem 26. Mai 2021 die Anforderungen der EU-MDR erfüllen. Mit der neuen Änderungsverordnung (EU) 2023/607, die die Übergangsfristen in der EU-MDR verlängert, wurde jedoch eine “Schonfrist” für implantierbare Sonderanfertigungen der Klasse III eingeführt, die eine Zertifizierung des Qualitätsmanagementsystems (QMS) durch eine Benannte Stelle benötigen würden (Art. 52 Abs. 8). Nach dem neuen EU-MDR-Artikel 120 Abs. 3 Ziffer f kann die Zertifizierung bis zum 26. Mai 2026 aufgeschoben werden, sofern der Hersteller bis zum 26. Mai 2024 einen formellen Antrag bei einer Benannten Stelle gestellt und die entsprechende schriftliche Vereinbarung bis zum 26. September 2024 unterzeichnet hat. In der Praxis wird diese neu eingeräumte “Schonfrist” vor allem denjenigen Herstellern zugute kommen, die bereits um den 26. Mai 2021 herum auf die EU-MDR umgestellt und das entsprechende Zertifikat der Benannten Stelle für ihr QMS erhalten hatten.
Obwohl Sonderanfertigungen nicht CE-gekennzeichnet sind, muss ein Hersteller von Sonderanfertigungen die Anforderungen der EU-MDR einhalten. Dazu gehören die Umsetzung von Anhang XIII und die Registrierung in EUDAMED als Akteur, auch wenn die UDI-Anforderungen nicht gelten. Die MDCG 2021-3 enthält Einzelheiten zu den verschiedenen Aspekten, die ein Hersteller von Sonderanfertigungen beachten muss.
Was ist eine “wesentliche Änderung” nach EU-MDR und IVDR?
Wenn das Produkt eine wesentliche Änderung des Produktdesigns oder der Zweckbestimmung erfährt, so verliert es den Legacy-Status und muss eine Zertifizierung durchlaufen sowie alle relevanten Anforderungen erfüllen.
“Änderung des Produktdesigns” — das mag einfach klingen, aber der Begriff wird in einem weiten Sinn verwendet.
Folgende Liste zeigt zusammengefasst die Änderungen, die in den Leitfäden genannt werden (MDCG 2020-3 und MDCG 2022-6 zur Bewertung der Signifikanz von Änderungen):
- Änderungen im Zusammenhang mit einer Korrekturmassnahme, es sei denn, sie wurden von der zuständigen Behörde am Sitz des Herstellers oder seines EC-REP geprüft und akzeptiert.
- Änderungen der Zweckbestimmung des Produkts, z.B. Erweiterungen, neue Anwender/Patientengruppen, neue Art der klinischen Anwendung.
- Änderungen der Produktauslegung, die den eingebauten Kontrollmechanismus, das Funktionsprinzip, die Energiequelle oder Alarmsysteme verändern, sowie alle Änderungen, die die Sicherheit oder Leistung beeinträchtigen und sich negativ auf das Risiko-Nutzen-Verhältnis auswirken können.
- Die meisten Softwareänderungen.
- Bestimmte Änderungen an Materialien oder Substanzen von Medizinprodukten oder IVD-Bestandteilen.
- Änderungen der Methode zur Endsterilisation oder des Verpackungsdesigns mit Auswirkungen auf den sterilen Zustand des Produkts.
Wenn das Produkt eine wesentliche Änderung am Produktdesign oder bei der Zweckbestimmung erfährt, so verliert es den Legacy-Status.
Weitere Einzelheiten zu wesentlichen Änderungen im Rahmen der EU-MDR finden Sie in unserem Blog-Artikel “Wesentliche Änderungen bei Medizinprodukten und IVD gemäss EU-MDR/IVDR”.
Welche Übergangsfristen und -bedingungen gelten für Legacy-Produkte?
Für Legacy-Produkte wurden die nachstehenden Übergangsfristen für die Umstellung der CE-Kennzeichnung von den früheren Richtlinien auf die EU-MDR bzw. IVDR gewährt:
Für Medizinprodukte mit einer gültigen Bescheinigung:
- 31. Dezember 2027 für Produkte der Klasse III und implantierbare Produkte der Klasse IIb (ausser Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Platten, Drähte, Stifte, Clips und Verbindungsstücke).
- 31. Dezember 2028 für andere Produkte der Klasse IIb, der Klasse IIa und der Klasse Is/m.
Für die Zwecke dieser Fristen gilt die (künftige) Einstufung gemäss Anhang VIII der EU-MDR.
Zu den Bedingungen, die in Art. 120 Abs. 3 Ziffer c der EU-MDR aufgeführt sind, gehören:
- Das Produkt entspricht weiterhin der Richtlinie 90/385/EWG bzw. der Richtlinie 93/42/EWG,
- kein unannehmbares Risiko für Gesundheit und Sicherheit,
- keine wesentlichen Änderungen am Produktdesign und der Zweckbestimmung,
- das QMS des Herstellers gemäss EU MDR Art. 10 Abs. 9 ist bis spätestens 26. Mai 2024 eingerichtet,
- Nachweis eines Antrags bei einer Benannten Stelle für den Übergang zur EU-MDR bis zum 26. Mai 2024 und Unterzeichnung der schriftlichen Vereinbarung mit der Benannten Stelle bis spätestens zum 26. September 2024.
Für Medizinprodukte, für die nach MDD keine Benannte Stelle erforderlich war (z.B. Produkte der Klasse I und des Anhangs XVI der EU-MDR) und für die dies nach EU-MDR erforderlich ist: bis zum 31. Dezember 2028.
Für Medizinprodukte mit einer vor dem 20. März 2023 abgelaufenen Bescheinigung wurden die gleichen Übergangsfristen gewährt wie für Medizinprodukte mit einer gültigen Bescheinigung (Art. 120 Abs. 2 EU-MDR), sofern eine der folgenden Bedingungen erfüllt ist:
- Es wurde bereits vor Ablauf der Bescheinigung eine schriftliche Vereinbarung mit der Benannten Stelle für den Übergang auf die EU-MDR unterzeichnet, oder
- eine zuständige Behörde hat eine Ausnahmegenehmigung gemäss Art. 59 Abs. 1 der EU-MDR erteilt, oder
- eine zuständige Behörde hat den Hersteller aufgefordert, das geltende EU-MDR-Konformitätsbewertungsverfahren gemäss Art. 97 Abs. 1 der EU-MDR durchzuführen.
Weitere Informationen über abgelaufene Bescheinigungen finden Sie weiter unten.
Wie bereits erwähnt, können implantierbare Sonderanfertigungen der Klasse III, die eine von einer Benannten Stelle ausgestellte QMS-Bescheinigung benötigen, bis zum 26. Mai 2026 ohne eine solche Bescheinigung auf dem Markt bleiben, sofern bis zum 26. Mai 2024 ein förmlicher Antrag bei einer Benannten Stelle gestellt und die schriftliche Vereinbarung mit der Benannten Stelle bis spätestens 26. September 2024 unterzeichnet wurde (Art. 120 Abs. Ziffer f der EU-MDR).
Für IVD mit einer gültigen Bescheinigung:
- 31. Dezember 2027 für Klasse D IVD.
- 31. Dezember 2028 für Klasse C IVD.
- 31. Dezember 2029 für Klasse B und Klasse A Steril IVD.
Für die Zwecke dieser Fristen gilt die (künftige) Einstufung gemäss Anhang VIII IVDR.
Zu den Bedingungen gehören (Art. 110 Abs. 3 Ziffer c IVDR):
- Das Produkt entspricht weiterhin der Richtlinie 98/79/EWG,
- kein unannehmbares Risiko für Gesundheit und Sicherheit,
- keine wesentlichen Änderungen am Produktdesign und der Zweckbestimmung,
- das QMS des Herstellers ist bis spätestens 26. Mai 2025 eingerichtet (Art. 10 Abs. 8 IVDR),
-
Nachweis eines Antrags bei einer Benannten Stelle für den Übergang zur IVDR und Unterzeichnung der schriftlichen Vereinbarung mit der Benannten Stelle, innerhalb der folgenden klassenspezifischen Fristen:
- Für Klasse D IVD: Antragstellung bis 26. Mai 2025 und schriftlichen Vereinbarung bis 26. September 2025.
- Für Klasse C IVD: Antragstellung bis 26. Mai 2026 und schriftlichen Vereinbarung bis 26. September 2026.
- Für Klasse B und Klasse A steril IVD: Antragstellung bis 26. Mai 2027 und schriftlichen Vereinbarung bis 26. September 2027.
Darüber hinaus wurden im Rahmen der Änderungsverordnung (EU) 2023/607 zur Entschärfung des Engpasses beim Übergang zur EU-MDR und IVDR die Abverkaufsbestimmungen sowohl in der EU-MDR als auch in der IVDR (d.h. im Wesentlichen ein Jahr nach Ablauf der jeweiligen Übergangsfrist für die Aktualisierung der CE-Kennzeichnung) gestrichen, so dass die Bestände an Legacy-Produkten bei den Händlern auf unbestimmte Zeit über das Ende der jeweiligen Übergangsfrist hinaus verkauft werden können.
Die nachstehenden Infografiken fassen die neuen Fristen im Rahmen der EU-MDR und IVDR zusammen:
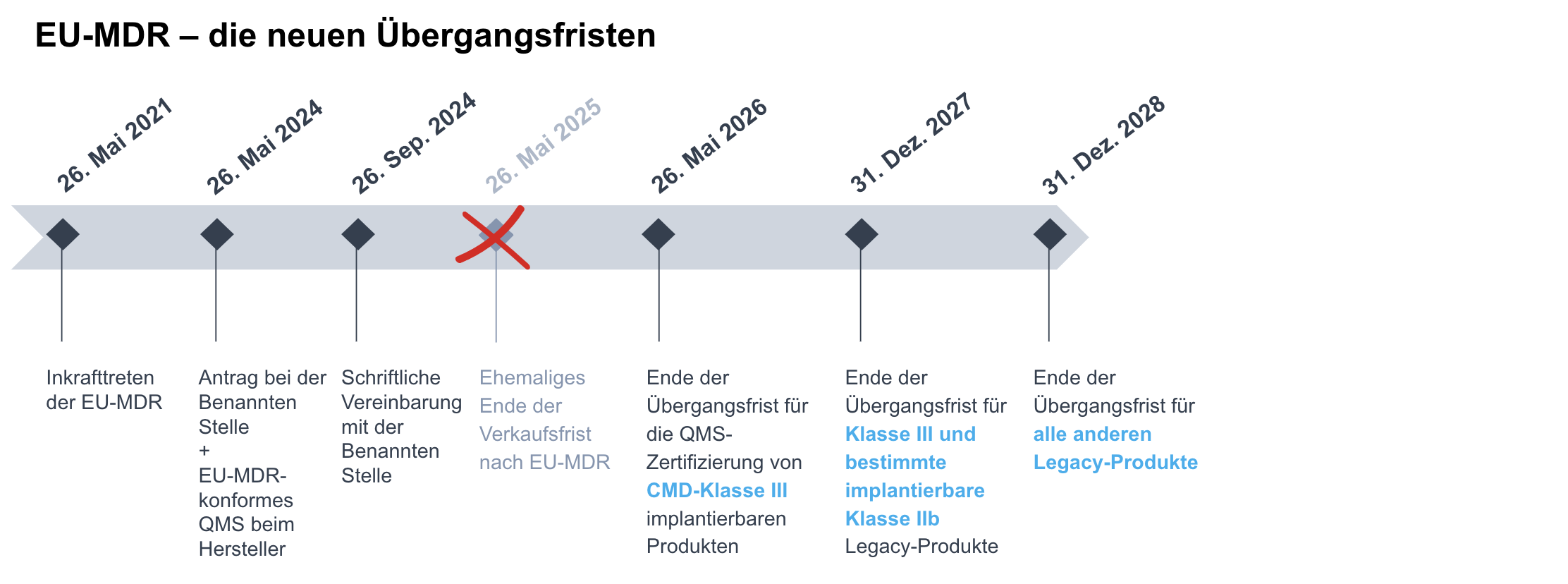
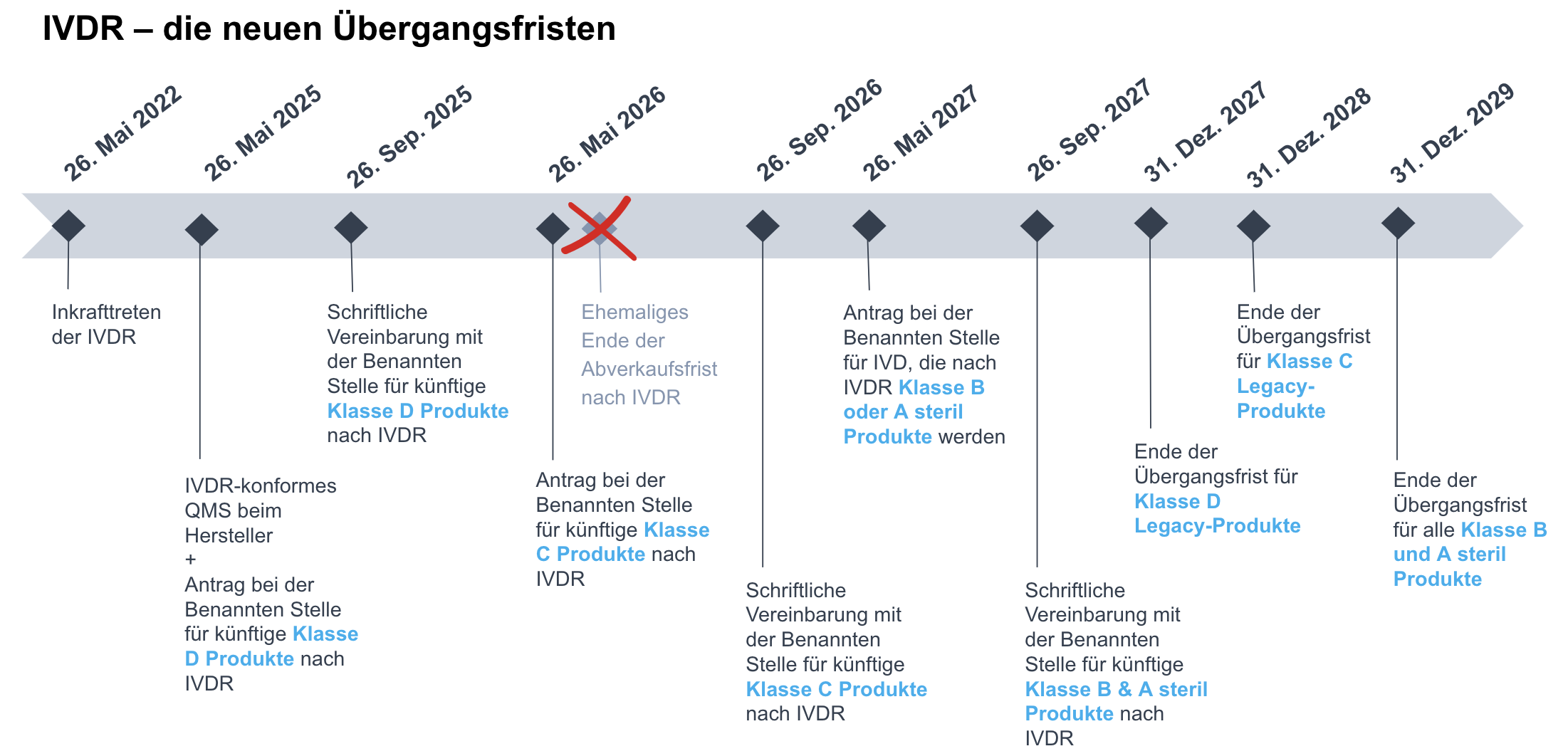
Um von den Übergansfristen für Legacy-Produkte zu profitieren, bedarf es eines Nachweises für die Erfüllung der Bedingungen. Dieser Nachweis wird erbracht durch:
- Eine Selbsterklärung des Herstellers, in der bestätigt wird, dass die Bedingungen für die Verlängerung erfüllt sind, mit Angabe des Enddatums der Übergangsfrist. Eine Vorlage für eine solche Selbsterklärung wurde von MedTech Europe zur Verfügung gestellt.
- Und ein Bestätigungsschreiben der Benannten Stelle über den Erhalt des Antrags des Herstellers auf Konformitätsbewertung und den Abschluss einer schriftlichen Vereinbarung. Die Benannten Stellen sind nicht verpflichtet, solche Schreiben auszustellen, da der Hersteller den Antrag auf Konformitätsbewertung und den Abschluss einer schriftlichen Vereinbarung mit einer Benannten Stelle auch auf andere Weise, z.B. durch eine Kopie der relevanten Unterlagen, nachweisen könnte.
Welche EU-MDR- oder IVDR-Anforderungen gelten für Legacy-Produkte?
Die Bestimmungen der EU-MDR und der IVDR, die für Legacy-Produkte gelten, sind in den entsprechenden Übergangsbestimmungen aufgeführt. Näher erläutert werden sie zudem in den Leitfäden MDCG 2021-25 (für die EU-MDR) und MDCG 2022-8 (für die IVDR), wo es nützliche Tabellen für die wichtigsten Artikel gibt. Kurz gesagt, die Pflichten, die für Legacy-Produkte gelten, betreffen die Überwachung nach dem Inverkehrbringen, die Marktüberwachung (d.h. durch die zuständigen nationalen Behörden), die Vigilanz sowie die Registrierung von Wirtschaftsakteuren und Produkten.
Alle Produkte, die auf dem Markt sind, müssen dem Stand der Technik entsprechen durch Sicherstellung der Konformität mit allen geltenden Normen und Vorschriften.
Überwachung nach dem Inverkehrbringen und Vigilanz
Die wichtigste Anforderung für Legacy-Produkte ist wahrscheinlich die Überwachung nach dem Inverkehrbringen. Um die MDR bzw. IVDR zu erfüllen, erfordert die Überwachung nach dem Inverkehrbringen eine systematische und proaktive Sammlung von Daten über die Sicherheit und Leistung des Produkts. Die EU-MDR und IVDR verlangen auch den sog. regelmässig aktualisierten Sicherheitsbericht (engl. PSUR).
|
Plan zur Überwachung nach dem Inverkehr-bringen (PMSP) |
MDR |
|
IVDR |
|
|
Bericht über die Überwachung nach dem Inverkehrbringen (PMSR) |
MDR |
|
IVDR |
|
|
Regelmässig aktualisierter Bericht über die Sicherheit (PSUR) |
MDR |
|
IVDR |
|
|
Plan für Folgemassnahmen nach dem Inverkehrbringen (PMCFP) |
MDR |
|
IVDR |
|
|
Bericht über Folgemassnahmen nach dem Inverkehrbringen (PMCFR) |
MDR |
|
IVDR |
|
|
Vigilanz-Meldungen |
Gemäss EU MDR Artikel 87 oder IVDR Artikel 82: Ernsthafte Bedrohung der öffentlichen Gesundheit: Nicht später als 2 Tage. Tod oder schwerwiegende Verschlechterung des Gesundheitszustands: Nicht später als 10 Tage. Andere schwerwiegende Vorfälle: Nicht später als 15 Tage. Jede Sicherheitskorrekturmassnahme im Feld (einschliesslich derjenigen, die im Ausland auftreten und Produkte betreffen, die im EWR bereitgestellt werden) Trendberichterstattung, gemäss EU MDR Artikel 88 oder IVDR Artikel 83. |
* Eine vollständige Überarbeitung der technischen Unterlagen gemäss den Anhängen II und III ist jedoch nicht erforderlich.
Beachten Sie, dass für “Altprodukte”, wie unter Wann gilt ein CE-gekennzeichnetes Produkt (Medizinprodukt oder IVD) als Legacy-Produkt? beschrieben, die Meldung schwerwiegender Zwischenfälle Pflicht ist. Eine solche Meldung erfordert eine spezielle EUDAMED-Registrierung des Altproduktherstellers, wie in Frage 5 des Leitfadens MDCG 2021-13 beschrieben.
Registrierung von Wirtschaftsakteuren und Produkten
Grundsätzlich kann die EU-Kommission die EUDAMED-Registrierung von Wirtschaftsakteuren und Produkten nicht vorschreiben, solange EUDAMED nicht voll funktionsfähig ist. Dies ändert sich nun mit dem Vorschlag, EUDAMED in einer schrittweisen Einführung, die bis Ende 2025 beginnen soll, verbindlich zu machen.
Die Legacy-Produkte selber müssen auch in EUDAMED registriert werden (siehe MDCG 2019-5). Wenn diese Produkte nicht über eine Basis-UDI-DI oder eine UDI-DI verfügen, müssen sie eine EUDAMED-DI (anstelle der Basis-UDI-DI) und eine EUDAMED-ID (anstelle der UDI-DI) haben. Wenn diese Produkte dann in die EU-MDR- oder IVDR-CE-Kennzeichnung überführt werden, sollten Verknüpfungen zwischen den jeweiligen Registrierungen hergestellt werden.
Verwandte Verpflichtungen der Wirtschaftsakteure
In Bezug auf Legacy-Produkte gelten die Anforderungen der EU-MDR oder der IVDR, die sich nicht auf Überwachung nach dem Inverkehrbringen, Marktüberwachung, Vigilanz, oder Registrierung von Wirtschaftsakteuren und Produkten beziehen, für Wirtschaftsakteure im Prinzip nicht. Die Leitfäden MDCG 2021-25 (zur EU-MDR) und MDCG 2022-8 (zur IVDR) vertreten diesbezüglich die Auffassung, dass folgende Kapitel für die entsprechenden Wirtschaftsakteure relevant sind:
- für Hersteller gemäss EU-MDR: Art. 10 Abs. 10 und Art. 10 Abs 12 bis 15;
- für Hersteller gemäss IVDR: Art. 10 Abs. 9 und Art. 10 Abs. 11 bis 10 Abs 14;
- für Bevollmächtigte: Art. 11 Abs. 3 Buchstaben c) bis g);
- für Importeure: Art. 13 Abs. 2 Unterabsatz 2, Art. 13 Abs. 4, Art. 13 Abs. 6 bis 8 und Art. 13 Abs. 10;
- für Händler: Art. 14 Abs. 2 letzter Unterabsatz und Art. 14 Abs. 4 bis 6.

Die EU-MDR/IVDR-Konformität erfordert bei der Überwachung nach dem Inverkehrbringen eine systematische und proaktive Sammlung von Daten zur Sicherheit und Leistung des Produkts.
Brauchen Hersteller von Legacy-Produkten einen PRRC?
Die für die Einhaltung der Vorschriften verantwortliche Person (PRRC für Person Responsible for Regulatory Compliance) ist eine Anforderung, die in Artikel 15 der EU-MDR und IVDR eingeführt wurde. Wenn Sie mehr über die Rolle des PRRC erfahren möchten, lesen Sie bitte Die Rolle des PRRC in der MDR, die auch auf die IVDR übertragen werden kann.
Artikel 15 ist nicht in den Übergangsbestimmungen zur EU-MDR und IVDR für Legacy-Produkte aufgeführt. Dies wird auch in den Leitfäden MDCG 2021-25 und MDCG 2022-8 bestätigt, in denen Artikel 15 als Beispiel für Bestimmungen angeführt wird, die nicht für Legacy-Produkte gelten.
Wann gelten Behandlungseinheiten und Systeme als “Legacy-Produkte”?
Systeme und Behandlungseinheiten gelten als Legacy-Produkte, wenn sie nur aus Legacy-Produkten bestehen und wenn für sie vor dem 26. Mai 2021 eine Erklärung gemäss Art. 12 der MDD erstellt wurde.
Sobald eine Komponente des Systems oder der Behandlungseinheit die CE-Kennzeichnung erhält, verliert das gesamte System bzw. Behandlungseinheit seinen Legacy-Status. Alle Legacy-Produkte, die in einem System oder einer Behandlungseinheit enthalten sind, fallen jedoch unter die Übergangsbestimmungen von Art. 120 Abs. 3 der EU-MDR bzw. Art. 110 Abs. 3 der IVDR.
Was ist bei Legacy-Produkten in der Schweiz speziell zu beachten?
Die Schweiz gilt seit dem 26. Mai 2021 als Drittland im Sinne der EU-MDR und seit dem 26. Mai 2022 im Sinne der IVDR. Die schweizerische Gesetzgebung wurde entsprechend angepasst. Sie erkennt aber die CE-Kennzeichnung unter der Schweizer Medizinprodukteverordnung (MepV, SR 812.213) bzw, unter der Schweizer In-vitro-Diagnostika-Verordnung (IvDV, SR 812.219) an. Somit haben auch “Legacy-Produkte” und “Altprodukte” unter der schweizerischen Gesetzgebung die gleiche Bedeutung und Auswirkung wie unter der EU-MDR und IVDR. Da die schweizerische Gesetzgebung im Wesentlichen auf die entsprechenden Bestimmungen der EU-MDR und IVDR verweist, müssen die jüngsten Aktualisierungen zur Ausweitung ihrer Übergangsbestimmungen in die MepV bzw. IvDV übernommen werden.
«Legacy-Produkte von Herstellern mit Sitz ausserhalb der Schweiz benötigen neu einen Schweizer Bevollmächtigten»
Kurz gesagt, die Anforderungen für Legacy-Produkte sind in der Schweiz die gleichen wie in der EU. Da die Schweiz nun jedoch ein Drittland ist, benötigen alle Legacy-Produkte von Herstellern mit Sitz ausserhalb der Schweiz neu einen Schweizer Bevollmächtigten. Darüber hinaus werden alle Schweizer Unternehmen, die Produkte aus dem Ausland in die Schweiz einführen und im Land weiterliefern, de facto zu Schweizer Importeuren und müssen neue Pflichten im Rahmen der MepV und IvDV erfüllen. Diese Anforderungen gelten unabhängig vom Status des Legacy-Produkts oder der Übergangsfrist des Produkts.
Zudem erwartet die zuständige schweizerische Behörde, Swissmedic, dass die Schweizer Bevollmächtigten und die Schweizer Importeure die Plausibilität der verlängerten Zertifikatsgültigkeit von Legacy-Produkten anhand der Selbstdeklarationen der Hersteller und der Bestätigungsschreiben der Benannten Stellen überprüfen. Für weitere Details siehe Frage (d) auf Seite 18 des Infoblatt zu den Pflichten der Wirtschaftsakteure von Swissmedic.
Wie Decomplix helfen kann
Decomplix berät Sie in allen regulatorischen und Qualitätsmanagement-Fragen rund um Medizinprodukte, mit besonderem Fokus auf den Schweizer Markt und die CE-Kennzeichnung.
Wir können Ihr Unternehmen dabei unterstützen, alle Anforderungen der EU-MDR oder IVDR zu erfüllen und die Konformität auf die effizienteste Art und Weise sicherzustellen. Wenn Sie Ihre Anliegen mit uns besprechen möchten, können Sie uns hier für ein unverbindliches Angebot zur CE-Kennzeichnung kontaktieren.
Darüber hinaus bieten wir Dienstleistungen als Schweizer Bevollmächtigter nach MepV und IvDV. Unsere Dienstleistungen umfassen Anleitungen und Checklisten, damit Sie die geltenden Anforderungen verstehen. Wenn Sie sich dafür interessieren, können Sie über dieses Formular Kontakt mit uns aufnehmen.
Weiterlesen
- Ist mein Produkt in Europa ein Medizinprodukt? Wie Sie feststellen, ob Ihr Produkt die CE-Zertifizierung für Medizinprodukte benötigt
- Wie man eine CE-Kennzeichnung für ein Medizinprodukt erhält
- Anforderungen an Medizinprodukte der Klasse I für Hersteller gemäss EU-MDR
- MDCG-Guidance zu wesentlichen Änderungen bei Medizinprodukten
- Schweizer Bevollmächtigter für Hersteller von Medizinprodukten



