
Is my product a medical device in Europe? How to determine if your product requires medical device CE marking
If you want to know whether your product is considered a medical device (or an in vitro diagnostic device, IVD) in the EU so to determine whether it needs medical device (or IVD) CE marking, read on. The answer to what may seem a simple question can be quite complex.
Replaces the version of 12.02.2019
Key takeaways:
- Accessories, components and even products with only aesthetic purposes can be viewed as medical devices. Make sure you can tell when.
- In assessing the regulatory category of your product, consider all the legal definitions and exclusions from scope of application in the EU MDR and IVDR for a correct qualification. Use the qualification decision flowchart in this article.
- For “borderline” products, do not trust your own interpretation. Consult the relevant official guidelines and even the European Court of Justice’s rulings, if needed.
Content:
What should be the starting point?
Determining the regulatory regime that applies to your product may pose a challenge. Some products are not easily categorized because they combine functionality corresponding to different regulatory regimes or because their “borderline” nature allows them to fall under one regime or another depending mostly on the claims made by the manufacturer. In such situations, the final decision is based on a case-by-case assessment, sometimes even going as far as a court ruling.
Consequently, you need to start with the intended purpose you assign to your product, which you should describe in sufficient detail to understand whether or not there is a medical action involved.
For example, a tweezer can be intended to remove glass or splinters from a wound (i.e. a medical purpose), for eyebrow plucking (i.e. a cosmetic purpose), or as an all-purpose tool which you can also use to remove fish bones (i.e. a general consumer product).
As a rule of thumb, if you make a medical claim for your product, chances are good that it is a medical device and taking a very close look at the legally binding definitions is indispensable.
How is the legal framework helpful for medical device qualification?
The current legal framework in the European Economic Area (EEA), Turkey, and Switzerland is based on the European Union’s Regulation (EU) No. 2017/745 on medical devices (EU MDR) and Regulation (EU) No. 2017/746 on in vitro diagnostic devices (IVDR). Both regulations apply in all EU Member States as they are, and are adopted in other EEA countries, Turkey and Switzerland to different degrees, through national legislation. For simplification, in this article we refer generically to the EU.
The EU MDR provides legally binding definitions of medical devices and accessories for medical devices, as well as exclusions from the scope of application of the regulation for demarcation between medical devices and other closely related products (e.g. pharmaceuticals, advanced therapy products, biological components, cosmetics or foodstuffs). In addition, EU MDR Annex XVI contains a list of products without an intended medical purpose to which the regulation also applies.
As to the IVDR, it sets forth the specific definitions for in vitro diagnostic devices (IVDs), which are in fact a subset of medical devices, as well as IVD accessories, and the corresponding exclusions of scope, which allow a differential qualification from non-IVD medical devices but also from general laboratory equipment or research only equipment. You can read more about CE marking of in vitro diagnostic devices under the IVDR here.
Taking both regulations together, the questions to consider for medical device (or IVD) qualification would be:
- Does the product fall under the definition of a medical device?
- Does the product fall under the definition of accessory for a medical device?
- Is the product listed in Annex XVI of the EU MDR?
- Is the product excluded from the scope of the EU MDR?
- Does the product fall under the definition of an IVD?
- Does the product fall under the definition of accessory for an IVD?
- Is the product excluded from the scope of the IVDR (and is it not an invasive specimen sampling product)?
which can be navigated through via the below qualification decision flowchart.
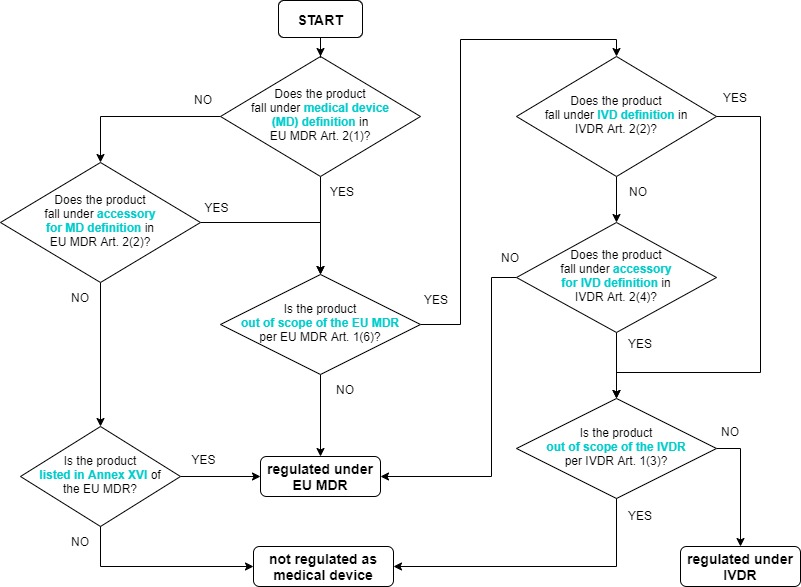
This means that the first legal definition you have to peruse is that of a medical device, in EU MDR Article 2 (1):
‘medical device’ means any instrument, apparatus, appliance,software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
- diagnosis, prevention, monitoring, prediction, prognosis, treatment, or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
- investigation, replacement, or modification of the anatomy or of a physiological or pathological process or state,
- providing information by means of in vitro examination of specimens derived from the human body, including organ, blood, and tissue donations,
and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
The following products shall also be deemed to be medical devices:
- devices for the control or support of conception;
- products specifically intended for the cleaning, disinfection or sterilisation of devices as referred to in Article 1(4) and of those referred to in the first paragraph of this point.
The definition is based on the intended use of your product (bringing you back to your medical claims), which is ultimately the decisive element of the qualification, and further nuanced by the principal mode of action, to allow differentiation from pharmaceuticals, which are the closest borderline products.
If your product does not have any of the medical purposes in the above definition, it may still be viewed as a device under the EU MDR, since accessories and Annex XVI products need also to be considered.
“Accessory” is a term that is legally defined both in the EU MDR and the IVDR.
- Accessories for medical devices mean, per EU MDR Article 2(2):
an article which, whilst not being itself a medical device, is intended by its manufacturer to be used together with one or several particular medical device(s) to specifically enable the medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the medical device(s) in terms of its/their intended purpose(s);
- And similarly, accessories for IVDs mean, per IVDR Article 2(4):
an article which, whilst not being itself an in vitro diagnostic medical device, is intended by its manufacturer to be used together with one or several particular in vitro diagnostic medical device(s) to specifically enable the in vitro diagnostic medical device(s) to be used in accordance with its/their intended purpose(s) or to specifically and directly assist the medical functionality of the in vitro diagnostic medical device(s) in terms of its/their intended purpose(s);
Therefore, you need to carefully review the items you sell in order to determine whether they fall under the above definitions or they are some type of convenience item that you do not intend to be used together with a medical device or IVD to specifically enable its use. Again, it is a matter of the item’s intended purpose.
And, to make things even more complicated, you need to consider device parts or components, too. These tend to be understood by manufacturers as spare or optional items that are shipped separately from the device they are intended to be used with. Under both the EU MDR and the IVDR, parts and components that significantly change the performance or safety characteristics or the intended purpose of a device are also considered devices.
Moreover, a new category of products has been introduced under EU MDR Annex XVI. These are mostly equipment and items for aesthetic purposes that were not previously regulated and must now conform to medical device requirements. They include:
- Contact lenses or other items intended to be introduced in/on the eye.
- Surgically invasive products for the purpose of modifying the anatomy or fixation of body parts (except tattooing products and piercings).
- Substances, combinations of substances, or items intended for facial or other dermal or mucous membrane filling (except those for tattooing).
- Equipment to reduce, remove or destroy adipose tissue (e.g. equipment for liposuction, lipolysis or lipoplasty).
- High intensity electromagnetic radiation equipment (e.g. infra-red, visible light and ultra- violet) intended for skin resurfacing, tattoo or hair removal or other skin treatment.
- Equipment intended for brain stimulation that apply electrical currents or magnetic or electromagnetic fields that penetrate the cranium to modify neuronal activity in the brain.
If your product has an in vitro application, meaning that its intended use revolves around the examination of body specimens (e.g. blood, saliva, urine), you want to consult the IVD definition in IVDR Article 2(2) to ensure you select the correct governing regulation. Note that:
- A product cannot be claimed to fall under the scope of the IVDR just by indicating ‘for in vitro diagnostic use’; it has to have a medical device purpose.
- Items meant to obtain body specimens invasively or from the body surface (e.g. lancets, swabs) are considered medical devices and are governed by the EU MDR.
- General laboratory equipment and research-only products are not considered IVDs and, thus, are viewed as general consumer products.
Accessories, components and even products for aesthetic purposes only might still require CE-marking as medical devices.
Last, if your product falls under the definition of a medical device in EU MDR Article 2(1) and/or the definition of a IVD in IVDR Article 2(2), it may be excluded from their scope and be governed by a different regulatory regime. For example, provided the product meets the corresponding legal definition:
- Pharmaceuticals are regulated under Directive 2001/83/EC. In practice, drug-device combination products and medical devices consisting of substances (e.g. ocular rinsing solutions) are not always easy to categorize.
- Advanced therapy products are regulated under Regulation (EC) No 1394/2007.
- Transplants, tissues/cells of human origin and their derivatives are regulated under Directive 2004/23/EC. However, medical devices (or accessories or Annex XVI products) manufactured using tissues/cells of human origin remain under the scope of the EU MDR.
- Cosmetics are regulated under Regulation (EC) No. 1223/2009.
- Foodstuffs are regulated under Regulation (EC) No. 178/2002.
With the above information in hand, you certainly realize that the qualification is only easy for very obvious medical devices. If that is not your case, we recommend that you continue reading.
“Borderline” products: how to navigate the uncertainty?
Despite the legal definitions and exclusions from the scope of application, some cases are still in-between regulations. For example: is a disinfectant a pharmaceutical, a medical device, or a biocide? Well, it depends on what it is intended to disinfect and how the disinfecting action is achieved.
There are a number of tools to navigate the uncertainty in the qualification of these products and to avoid the common mistake of trusting your own interpretation of a “borderline” situation without further looking for an official regulatory position.
One of the most comprehensive guidelines under the former EU legislation was the European Commission’s Manual on Borderline and Classification, last updated in May 2019. It compiled examples of “borderline” products and the conclusions agreed and shared by the EU regulators. A corresponding Manual on Borderline and Classification under the EU MDR and IVDR has been published in September 2022, as part of the work conducted by the Borderline & Classification Working Group with the Medical Device Coordination Group (MDCG). This first version contains only a few cases and will be further populated as more decisions are made. Like for any official guidelines, the views presented in this manual are not legally binding and only the European Court of Justice (ECJ) is entitled to issue an authoritative interpretation of medical device qualification matters, as any other aspect of EU legislation.
The new Manual on Borderline and Classification is structured in 2 sections, separately covering the EU MDR and the IVDR, with sub-sections relative to qualification and classification issues.
The EU MDR qualification sub-section encompasses 9 chapters divided into different sections, highlighting where difficulties lie:
- Chapter 1.1.1: Borderline between medical devices and IVDs.
This chapter will include the cases where the conclusion is that the product should qualify as a medical device. Products that should qualify as IVDs will be discussed in chapter 2.1.1. - Chapter 1.1.2: Borderline between medical devices and medicinal products, including advanced therapy medicinal products (ATMPs).
A separate MDCG guidance has also been published (see below). - Chapter 1.1.3: Borderline between medical devices and biocides.
- Chapter 1.1.4: Borderline between medical devices and substances of human origin.
- Chapter 1.1.5: Borderline between medical devices and cosmetic products.
- Chapter 1.1.6: Borderline between medical devices and food.
- Chapter 1.1.7: Borderline between medical devices and personal protective equipment.
- Chapter 1.1.8: Borderline between medical devices and general consumer products.
This chapter covers products that may or may not have a medical purpose. It is probably the most useful chapter for manufacturers of consumer products who make or plan to make medical claims (e.g. biofunctional clothes). Note that, the products listed in EU MDR Annex XVI are considered medical devices despite their lack of medical purpose. - Chapter 1.1.9: Other medical device borderlines.
The IVDR qualification sub-section encompasses only 3 chapters:
- Chapter 2.1.1: Borderline between IVDs and medical devices.
Note that a guideline had been issued still under the former Directive 98/79/EC (IVDD), MEDDEV 2.14/1, with solid regulatory basis for the demarcation between IVDs and non-IVDs that can still be of use in the absence of IVDR-specific MDCG guidance documents and/or “borderline” cases described in this Manual. - Chapter 2.1.2: Borderline between IVDs and general laboratory equipment.
A separate MDCG guidance has also been published (see below). - Chapter 2.1.3: Other IVD borderlines.
The new Manual also includes sections of Classification issues, structured per EU MDR and IVDR classification rules.
The MDCG had previously published specific guidance documents that need to be read in conjunction with the new Manual on Borderline and Classification, if they concern your product:
- Published in October 2019, MDCG 2019-11 groups qualification and classification guidance on medical device software (MDSW). This concerns both software that is embedded in a hardware medical device and standalone software.
- Published in April 2022, MDCG 2022-5 is an MDR-specific guidance document on the borderline cases between medical devices and medicinal products.
In addition, the MDCG guidance documents on medical device classification rules can also be useful sources of regulatory opinion as they contain numerous examples. These are: MDCG 2021-24 for non-IVD medical devices and MDCG 2020-16 for IVDs.

Use the tools available to navigate the uncertainty associated with the qualification of “borderline” products.
In brief, if you suspect that your product is a medical device, we advise you to consult the legal framework as well as the available official guidelines mentioned above, and even the ECJ’s rulings that are publicly available from the InfoCuria Case-law database.
If doubts persist and you believe that your product could be a medical device of the lowest risk class, you can send a request for support to your national competent authority, bearing in mind that competent authorities do not offer consultancy services. If a higher risk class comes into question, the first request for support should be addressed to a Notified Body, which could still involve the competent authority, if necessary.
You can find out more about medical device risk classification here.
How Decomplix can help
Decomplix provides expert assessment of your situation and a complete roadmap to obtaining a CE mark for your medical device. You can learn more about our services here.



