
Swiss medical device importers – regulatory requirements
Are you unclear about the requirements for importing medical devices (incl. in-vitro diagnostic devices, IVDs) into Switzerland? Have you heard about “virtual” Swiss importers and wonder whether this service can help you? Do you sell medical equipment or medical supplies in Switzerland and wonder whether you are viewed as a Swiss Importer? Here is all you need to know about it.
Replaces version of 27.06.2022
Key takeaways:
- Any Swiss-based company placing medical devices from abroad on the Swiss market becomes a Swiss Importer.
- The concept of “placing on the market” is essential. This turns pharmacies, opticians, supermarkets, online shops or other dispensing outlets into Swiss importers, too.
- There is no such thing as a “virtual” medical device importer. Be careful when choosing your importer in Switzerland.
- It is important to become familiar with your compliance duties as a Swiss Importer. If you think you need expert assistance, please contact us.
Content:
Why is a Swiss Importer suddenly needed for medical devices, incl. in-vitro diagnostic devices (IVDs)?
Importing CE-marked medical devices (including IVDs) into Switzerland used to be indistinguishable from importing them into any other EEA country, because the former European Directives 93/42/EEC concerning medical devices (MDD), 90/385/EEC on active implantable medical devices (AIMDD), and 98/79/EC on in vitro diagnostic medical devices (IVDD) were fully adopted under the former Swiss Medical Device Ordinance (MedDO, SR 812.213), based on the Mutual Recognition Agreement (MRA) between Switzerland and the EU. This is no longer the case.
Since 26 May 2021, date of application of the new Regulation (EU) 2017/745 on medical devices (EU MDR), Switzerland is considered a third country to the effects of the EU MDR. On the same date, Switzerland enacted a revised version of its Medical Device Ordinance, SR 812.213 (MedDO), which introduced Swiss-specific requirements to account for the absence of an updated MRA, some of which impact Swiss-based companies importing medical equipment or medical supplies. The same has subsequently happened for IVDs through the new Swiss In-vitro Diagnostic Medical Device Ordinance (IvDO) that came into effect on 26-May-2022, simultaneously to the date of application of the new Regulation (EU) 2017/746 on IVDs (IVDR).
«Switzerland is considered a third country to the effects of the EU MDR.»
In order to revert the situation, the MRA would need to be updated, which is unlikely to happen anytime soon as the EU Commission will not negotiate or update the MRA until an agreement between both parties has been established. Switzerland’s decision to not sign and to stop the process of the institutional agreement (InstA) unilaterally has wide-ranging and long-term consequences. The current political situation in Switzerland makes it thus highly unlikely that there will be a solution in the foreseeable future.
Without an updated MRA, Switzerland will remain as a “third country” to the effects of the EU MDR and IVDR, and EEA countries will remain as “third countries” under the Swiss legislation on medical devices and IVDs.
What is a medical device importer and who can endorse this role in Switzerland?
Per the definition in Article 4 §1(h) of the MedDO and Article 4 §1(g) of the IvDO, a medical device importer in Switzerland corresponds to:
“any natural or legal person established within Switzerland that places a device from a foreign country on the Swiss market”.
This means that any Swiss-based entity who places devices from abroad on the Swiss market from a foreign country becomes an importer. In other words, an entity becomes the Swiss Importer “by action” as opposed to by explicit designation by the foreign manufacturer.
In practice, this restricts the possibility of a foreign manufacturer to ship goods directly to distributors or retailers, if such companies are not ready to endorse the duties of a Swiss Importer. Also, pharmacies, supermarkets, online shops and other dispensing outlets who sell devices received directly from abroad are considered to be Swiss Importers.
It is important to bear in mind that, as defined in Article 4 §1(b) of the MedDO/IvDO, “placing on the market” means the first making available on the Swiss market, which is in turn defined in Article 4 §1(a) of both ordinances as any supply of a device (other than an investigational device) for distribution, consumption or use within Switzerland, and does not correspond to bringing a medical device across the Swiss border, which is a common misconception.
Swissmedic further elaborates the concept of supply in chapter 3 of its InfoSheet on the Obligation of Economic Operators, as follows:
“The making available of a product supposes an offer or an agreement (written or verbal) between two or more legal or natural persons for the transfer of ownership, possession or any other right concerning the product in question after the stage of manufacture has taken place8. The transfer does not necessarily require the physical handover of the product. This transfer can be for payment or free of charge.”
If such “transfer of ownership, possession or any other right” is undertaken by a given Swiss company, then this entity is the Swiss Importer.

The Swiss Importer role is determined “by action”. There is no such thing as a “virtual” or “designated” Swiss importer.
Note that, from a regulatory requirements standpoint, “parallel imports” (i.e. shipments from a foreign entity that is not the manufacturer) are not viewed differently from direct imports from the foreign manufacturer. In either case, the entity based in Switzerland that would place the devices on the Swiss market would be a Swiss importer.
What are the duties of the Swiss Importer? Can these be delegated?
The duties of a Swiss importer under the MedDO/IvDO are relatively straightforward as they mirror those of an importer under Article 13 of the EU MDR/IVDR. Pursuant to Article 53 of the MedDO and Article 46 of the IvDO, the duties of a Swiss Importer can be clustered into the following sets of activities:
Verification of compliance
Prior to “placing on the Swiss market” a device in accordance with Article 53 §1 of the MedDO and Article 46 §1 of the IvDO, the Swiss Importer shall verify that:
- the device carries the CE marking,
- the Declaration of Conformity has been drawn up,
- the manufacturer has designated a Swiss Authorised Representative,
- the device is labelled according to the MedDO/IvDO, and
- where applicable, a UDI has been assigned by the manufacturer.
Declarations of Conformity and, where applicable, Notified Body certificates shall be retained by the Swiss Importer for 10 years (15 years for implantable devices) after the last device was placed on the market, per EU MDR/IVDR Article 13(9).
Moreover, per Article 21 §2 of the MedDO and Article 17 §2 of the IvDO, the Swiss Importer is expected to be able prove that the conformity assessment has been conducted and that the device is compliant. This places significant burden on any Swiss importer, particularly for the verification of extended validity of EC Certificates for “legacy” devices under the MDD/AIMDD. Swissmedic’s expectation is for Swiss importers to check the plausibility of:
- the manufacturer’s self-declaration under EU MDR Art. 120(3c),
- the Notified Body’s confirmation letter stating that the manufacturer has lodged an application for EU MDR certification.
Per Article 53 §3 of the MedDO and Article 46 §3 of the IvDO, Swiss Importers who consider or have reason to believe that a device is not in conformity with the MedDO/IvDO, must not place the device on the market until it has been brought into conformity.
In the event of non-conformities, a process must be in place to inform the manufacturer and its Swiss Authorised Representative.
Suitable storage and transportation conditions
While a device is under the responsibility of the Swiss Importer, per EU MDR/IVDR Article 13(5), the storage/transportation conditions should not jeopardize the device compliance with the General Safety and Performance Requirements of the EU MDR/IVDR (or with the Essential Requirements for “legacy devices” still under the MDD/AIMDD/IVDD). In addition, where available, the Swiss Importer shall comply with any storage and transportation conditions set by the manufacturer.
Device traceability
Per MedDO Article 64 and IvDO Article 57, the Swiss Importer shall cooperate with the manufacturer or its Swiss Authorised Representative to ensure appropriate device traceability. This includes disclosing to Swissmedic on request the identity of economic operators from whom they have acquired a medical device and the economic operators or customers to whom they have supplied a medical device, per Article 47c of the Swiss Therapeutic Product Act (TPA). This duty of disclosure continues to apply for at least 10 years (15 years for implantable devices) from the date the device was procured or supplied.
Registry of complaints, non-conforming products, recalls and withdrawals
By analogy, per EU MDR/IVDR Article 13(6), the Swiss Importer shall maintain a register of complaints, of non-conforming devices and of recalls and withdrawals, and provide the manufacturer, its Swiss Authorised Representative, and the distributors with any information requested by them, in order to allow them to investigate complaints.
Communication
By analogy, per EU MDR/IVDR Article 13(8), the Swiss Importer shall immediately forward any complaints and incidents from the field to the foreign manufacturer and to its Swiss Authorised Representative. This applies also in the case of parallel imports, where the Swiss importer might not have a direct commercial relationship with the foreign manufacturer. Swiss Importers shall also ,where a non-conforming device presents a serious risk, immediately inform Swissmedic, giving details, in particular, of the non-compliance and of any corrective action taken, in accordance with EU MDR/IVDR Article 13(7), by analogy.
Cooperation
The Swiss Importer shall cooperate with Swissmedic, the Notified Body (where applicable), the manufacturer and its Swiss Authorised Representative, in case of corrective actions to bring a device into conformity (incl. recalls and withdrawals), per EU MDR/IVDR Article 13(10), by analogy.
Registration with Swissmedic (CHRN)
Also, per Article 55 §1 of the MedDO and Article 48 §1 of the IvDO, the Swiss Importer needs to register for its economic operator role with Swissmedic, which involves obtaining a Swiss Registration Number (CHRN). For this registration, the time of the first placing on the market matters:
- If the importer starts placing on the Swiss market either a non-IVD device for the first time after 26 May 2021 or an IVD for the first time after 26 May 2022, the registration must be done within 3 months.
- If the importer had already placed EU MDR-compliant devices on the Swiss market for the first time before 26 May 2021, they should have registered before 26 November 2021.
- If the importer had already placed IVDR-compliant IVDs on the Swiss market for the first time before 26 May 2022, they should have registered before 26 November 2022, per IvDO Article 88.
- If the importer had already placed on the Swiss market either “legacy” non-IVDs for the first time before 26 May 2021 or “legacy” IVDs before 26 May 2022, there is no obligation to register.
Note that for Swiss Importers who deal with both IVDs and non-IVD devices, the registration with Swissmedic is needed only once. Any subsequent changes to the Swiss Importer’s information notified (e.g. change in address) shall be submitted to Swissmedic within 1 week, per Article 55 §2 of the MedDO and Article 48 §2 of the IvDO.
Indication of Swiss importer’s particulars
Per Article 53 §2 of the MedDO and Article 46 §2 of the IvDO, the Swiss importer’s particulars must appear on the device, its packaging or an accompanying document (which can be the invoice, a delivery note, a guarantee or similar). See also: Do foreign manufacturers need to change the device labelling to mention the Swiss Importer?
All the above duties must be officially endorsed by the Swiss Importer, i.e. the Swiss Importer remains responsible and accountable before Swissmedic for all its obligations under the MedDO/IvDO. However, since there is no specific restriction in the MedDO/IvDO or the EU MDR/IVDR, related operational activities could be delegated, under the control of the Swiss importer. For example, warehousing activities could be delegated to a Swiss-based logistics provider or regulatory activities (e.g. “remote” verification of incoming goods) could be delegated to another entity within a big corporation or to a regulatory consultancy company. Note that delegation of regulatory activities does not mean that the company undertaking such activities can be considered to be the Swiss importer.

Any delegation of activities would need to be documented in a quality agreement between the parties involved.
Note also that Question 16 in guidance document MDCG 2021-27 on the roles of importers and distributors under the EU MDR/IVDR explicitly forbids the delegation of legal responsibility from one importer to another, because importers have an essential duty in overseeing the supply chain and ensuring traceability.
Any delegation of activities would need to be documented in a quality agreement between the parties involved.
As a foreign medical device manufacturer, can I designate a single Swiss importer, who takes on the corresponding duties in a “virtual” manner?
As indicated earlier, a Swiss entity becomes a Swiss importer by action. Therefore, there is only commercial but no regulatory designation of Swiss importers.
A few examples:
- A regulatory service provider offers services as “virtual” Swiss importer for foreign manufacturers. The company undertakes some or all regulatory activities, e.g. keeping the register of complaints, non-conformities and recalls/withdrawals, or communicating with Swissmedic, or remotely verifying compliance for each device placed on the market on behalf of the entity that actually imports and places such device on the Swiss market.
In this case, the regulatory service provider cannot be considered to be the Swiss importer and should not register with Swissmedic as the Swiss importer.
This service provider is merely being “delegated” some operational tasks. - A foreign manufacturer “designates” its Swiss subsidiary as the Swiss importer but actually ships the devices directly to various distributors or retailers located in Switzerland. Two cases are possible:
- If the “transfer of ownership” of the devices brought into Switzerland happens between the foreign manufacturer and its subsidiary, and then from the subsidiary to the Swiss distributor or retailer, the subsidiary can be considered to be the Swiss importer even if the goods are never shipped to or stored by the subsidiary.
- If the “transfer of ownership” of the devices brought into Switzerland happens only between the foreign manufacturer and the Swiss distributor or retailer, the subsidiary cannot be considered to be the Swiss importer. Instead, the Swiss distributor or retailer becomes the Swiss importer.
- A foreign manufacturer “designates” a Swiss entity as its single importer but then ships and sells the devices directly to hospitals. Two cases are possible:
- Direct shipment from foreign manufacturers to hospitals who are the end users is not considered to be “placing on the market” and, thus, a Swiss importer (and CH-REP, read more …) is not needed. Therefore, if the hospital imports the devices for its own use, it is not viewed as a Swiss importer.
- If the hospital further sells (or otherwise transfers the ownership of) the device, then it becomes a Swiss importer.
Is a Swiss Importer required in all circumstances? What about medical device software?
A Swiss Importer is not involved in the case of transactions concerning devices that are not “placed on the market” in Switzerland, as defined in Article 4 §1(b) of the MedDO/IvDO. This is the case for:
- Investigational devices.
- Devices imported directly from abroad to the end user, whether a healthcare professional, a healthcare institution, or the patient. This is considered to be direct use in Switzerland. In the case of healthcare professionals, such use is governed by Article 70 of the MedDO (or Article 63 of the IvDO), which places the responsibility for the conformity of the device on the healthcare professional. More details are available from Swissmedic’s Information Sheet on Procurement of medical devices in healthcare Institutions.
- Devices that had been already transferred or supplied by the Swiss Importer to the distributor before the MedDO/IvDO entered into force and are still at the distributor’s warehouse. Such devices are considered to have been placed on the market in Switzerland under the old legislation. Therefore, the Swiss Importer does not need to be stated retrospectively.
As to medical device software, neither the MedDO/IvDO nor the EU MDR/IVDR differentiate software from hardware medical devices that are placed on the Swiss market in terms of requirements applicable to importers.
Obviously, duties relative to appropriate storage and transportation conditions would have to be understood as those relative to software deployment, since there is no physical product. However, all other duties would apply as they are. Therefore, a Swiss Importer would be any Swiss-based entity who places on the market medical device software in Switzerland, e.g. by making it available to end users from its own platform.
Can the Swiss Authorised Representative also be the Swiss Importer?
The Swiss Authorised Representative and the Swiss Importer are different economic operator roles with different regulatory duties under the MedDO and the IvDO, and these roles tend to be undertaken by different types of companies, as further explained below.
However, both roles could be combined under the same entity. A company that chooses to act both as Swiss Authorised Representative and Swiss Importer shall register with Swissmedic, under Article 55 of the MedDO and Article 48 of the IvDO, separately for each of these two separate economic operator roles, and shall obtain two separate Swiss Registration Numbers (CHRN) from Swissmedic.
Even if the Swiss Importer’s duties involve compliance verification and notification requirements, which could be considered akin to those applicable to the Swiss Authorised Representative, the obligations of a Swiss Importer also cover warehousing and transportation activities, including the necessary quality management system.
Whereas a Swiss Importer could take on the additional role of Swiss Authorised Representative, provided it had sufficient in-house regulatory resources, the opposite is rarely the case since importation entails, in addition to regulatory compliance with the MedDO/IvDO, customs clearance, logistic, and tax-related activities and companies offering Swiss Authorised Representative services tend to be regulatory consultancy companies without competence in those domains. The role of a Swiss Importer is naturally easier for an existing Swiss distributor.
Read more about the role of a Swiss Authorised Representative.

Relabelling/repackaging requirements are the same as those under the EU MDR/IVDR.
Can a company simultaneously act as Swiss distributor and Swiss Importer?
No.
The Swiss importer and Swiss distributor are separate regulatory roles under the MedDO/IvDO.
These roles should not be mistaken with the concept of distributing goods, in the sense of shipping or selling. Of course, both importers and distributors can and do ship or sell devices but these activities are not the determining factor.
The Swiss importer brings medical devices from abroad (either from the foreign manufacturer or from a foreign distributor) and “places” them on the Swiss market, i.e. the device transfer or supply from the Swiss importer to a Swiss distributor or healthcare facility or the end user. This corresponds to the red arrow in the below infographic from Swissmedic.
The Swiss distributor only “makes available” devices on the Swiss market, i.e. further supply to other Swiss distributors or retailers, or to a healthcare facility or the end user. This corresponds to either the transparent or the green arrow in the below infographic from Swissmedic.
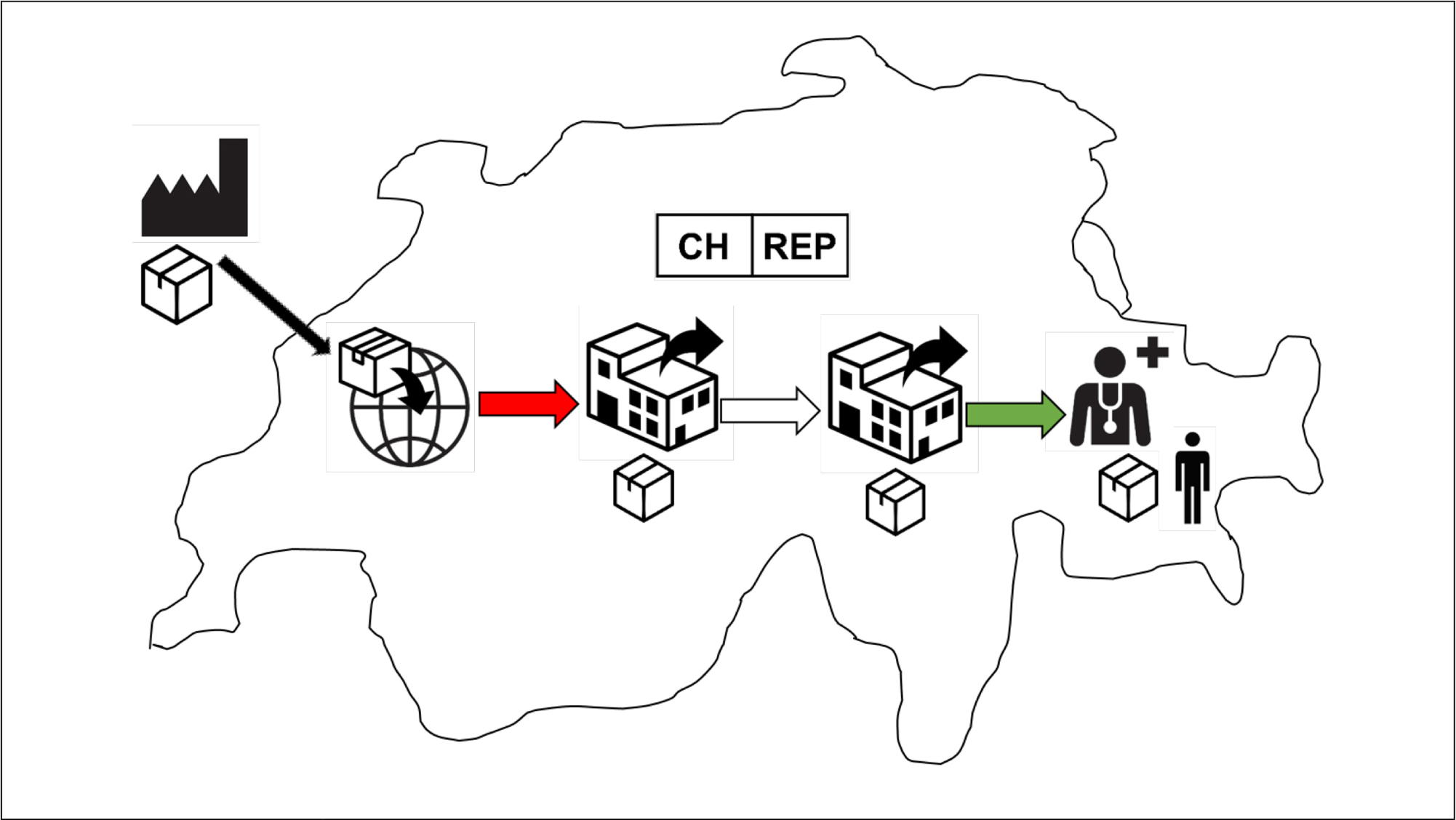
The Swiss importer and Swiss distributor are separate regulatory roles under the MedDO/IvDO.
Do foreign manufacturers need to change the device labelling to mention the Swiss Importer?
According to Article 53 §2 of the MedDO and Article 46 §2 of the IvDO, the name and address of the Swiss Importer must appear on the device, on the packaging or in a document accompanying the device. Examples of documents accompanying the device: delivery note, warranty certificate, customs documents, invoice, a sticker on the packaging or instructions for use.
Swissmedic has provided additional details regarding the mandatory indication of Swiss Importer’s particulars in chapter 6 of its Information Sheet on the Obligations of Economic Operators. The “document accompanying the device” may be attached to or separate from the device. Even if it does not necessarily have to reach the end user, Swissmedic clearly indicates that the Swiss importer must be clearly identifiable along the supply chain, even when there is no distributor. The purpose of the information is a quick and clear identification of the economic operators responsible for the devices at hand, e.g. for the implementation of product recalls, for the reporting of incidents, for notifications of dangerous products or non-conformities and in the context of enforcement.
«Swiss importer must be clearly identifiable along the supply chain.»
In the absence of more specific guidance from Swissmedic, Decomplix recommends the use of the term “CH IMPORTER” in German, French and Italian, as follows : “CH IMPORTEUR / IMPORTATEUR / IMPORTATORE”, and to avoid the use of the importer symbol of ISO 15223-1, which could be mistaken for EU importer.
According to Article 16 §2 of the MedDO and Article 15 §2 of the IvDO relative to language requirements that apply to labelling, labels need to be translated into German, French and Italian. However, Decomplix considers that it is not necessary to translate the city in the Swiss Importer’s address into other Swiss languages as addresses within Switzerland are driven by the postal code.
Is relabelling and repackaging by Swiss Importers allowed?
The requirements for entities that relabel and/or repackage devices under the MedDO/IvDO are the same as those under the EU MDR/IVDR.
Different situations are possible, which are described below.
1. The Swiss Importer undertakes relabelling or repackaging as subcontractor, on behalf and under the control of the legal manufacturer.
Chapter 7 of Swissmedic’s Information Sheet on the Obligations of Economic Operators clarifies that Swissmedic bases its interpretation of relabelling and repackaging requirements on the European practice and specifically on the guidance documents issued by the Medical Device Coordination Group (MDCG). This means that MDCG 2021-26 (Q&A on repackaging and relabelling activities) needs to be considered. Section 2 of this guidance document indicates that:
“Article 16(2), (3) and (4) of the Regulations do not apply to operators subcontracted by the manufacturer (that may also qualify as importers or distributors), who also carry out relabelling and/or repackaging activities on behalf and under the control of the manufacturer.”
In such a case, a specific quality agreement for the subcontracted activities is expected to be in place between the manufacturer and the Swiss Importer.
2. The Swiss Importer undertakes relabelling or repackaging on its own account, without a “private label” agreement.
Two options are possible:
The Swiss Importer acts or intends to act as “Own Brand Labeller (OBL)”, a concept that implies that the OBL is supplied with the final device by its supplier, usually known as “Own Equipment Manufacturer (OEM)”, and therefore modifies the packaging/labelling to appear as the entity placing the device on the market.
«OBLs are considered manufacturers under the EU MDR/IVDR.»
Per question IV.5 in guidance document MDCG 2019-6 (Q&A requirements relating to Notified Bodies), OBLs are considered manufacturers under the EU MDR/IVDR and must comply with the corresponding requirements for manufacturers, which includes but is not limited to having:
“full and permanent access to the technical documentation; (ability for) post-market surveillance including post market clinical follow-up; sufficient technical competence; and control of the quality system (control of the design, manufacture and/or final verification and testing of the devices).”
The Swiss Importer does not intend to act as OBL. The activities must then be carried out within Article 16 (2) of the EU MDR/IVDR. And, under this article, only the following few activities are allowed: providing device information ( including undertaking translations) and/or repackaging the device to the extent necessary to market that device in Switzerland. Note that MDCG 2021-26 considers as “necessary” the need to supply a number of devices different from the number of devices supplied in the original packaging by the manufacturer.
In such cases, per Article 16 (3) & (4) of the EU MDR/IVDR, which are pointed to by Article 53 §4 of the MedDO and Article 46 §4 of the IvDO, respectively, the Swiss Importer must:
- Indicate on the device, its packaging or an accompanying document, the activity carried out,
- Have a quality management system that includes procedures to ensure accurate and up-to-date translations, and that relabelling/repackaging activities are carried out under appropriate conditions,
- Obtain a certificate from a Notified Body attesting that the quality management system complies with the requirements, and
- Inform Swissmedic and the manufacturer of such activities at least 28 days prior to making the relabelled/repackaged device available on the market.
3. The Swiss Importer undertakes relabelling or repackaging on its own account, with a “private label” agreement
Although the concept of “private label” does not exist either in the EU MDR/IVDR, in practice, it can be somewhat considered to be covered by Article 16 (1)(a), since the importer could make available on the market a device under its own name, provided an agreement with the legal manufacturer has been established whereby the legal manufacturer is identified as such on the label and is responsible for meeting the requirements placed on manufacturers under the EU MDR/IVDR.
This possibility is also foreseen in the definition of “manufacturer” under the MedDO (and the IvDO):
“manufacturer means a natural or legal person who manufactures or fully refurbishes a device or has a device designed, manufactured or fully refurbished, and markets that device under its name or trademark; this definition is subject to the clarifying explanations and exceptions in Article 16 paragraphs 1 and 2 of Regulation (EU) 2017/7457 on medical devices (EU-MDR)”.
How can Decomplix help?
Decomplix offers consultancy services in all regulatory and quality assurance matters relative to medical devices, with particular focus on the Swiss market. We support your company in finding an appropriate solution in your existing distribution chain and help Swiss importers navigate the new requirements and ensure compliance in the most efficient manner.
In addition, we offer Swiss Authorised Representative services under both the MedDO and the IvDO, since May 2021, and already represent numerous foreign manufacturers. The services include a mandate contract, detailed step-by-step instructions and checklists for your understanding of the applicable requirements.
Further reading
- Swiss Authorised Representatives for Medical Device Manufacturers – Decomplix
- Which MDR requirements apply to distributors of medical devices?
- “Legacy” medical devices and IVDs under EU legislation
- EU MDR language requirements — what manufacturers and distributors need to know – Decomplix
- How the medical device regulation system enhances patient safety



