
Post-market Surveillance (PMS) for medical devices under EU MDR and IVDR
Post-market Surveillance (PMS) for CE-marked medical devices is not a new concept brought by the EU MDR or IVDR and, yet, many actors still misunderstand the purpose, requirements, and connection with other post-market activities. More alarming, PMS does not seem to be a priority for manufacturers, who rather place their efforts in pre-market activities.
If you are unclear as to how to proceed or you are interested in what has changed from the former legislation (MDD, AIMDD, IVDD), read on.
Replaces the version of 03.10.2019
Key takeaways
- All device manufacturers must establish a PMS system in compliance with the EU MDR or IVDR, even for legacy devices. To that end, the PMS procedure is the first step.
- The PMS Plan under the EU MDR or IVDR is not just a table that cross-references other QMS procedures. The appropriate methods and tools must be described, as explicitly required in Annex III of both the EU MDR and IVDR.
- Make sure you have read and understood the guidance document MDCG 2022-21, before you start preparing PMS Plans and PSURs, but also PMS Reports.
- The extent and frequency of the PMS activities are labor-intensive. Make sure you have the necessary resources, which can be outsourced.
Contents
What is PMS under the EU MDR and IVDR?
Post-market Surveillance (PMS) under the new Regulations (EU) No. 2017/745 on medical devices (EU MDR) and 2017/746 on in-vitro diagnostic devices (IVDR) is the systematic process that manufacturers must set up to gain experience relative to their medical devices (incl. IVDs) placed on the market and to act upon it.
The same definition of PMS is provided for in EU MDR Art. 2(60) and IVDR Art. 2(63), namely:
“all activities carried out by manufacturers in cooperation with other economic operators to institute and keep up to date a systematic procedure to proactively collect and review experience gained from devices they place on the market, make available on the market or put into service for the purpose of identifying any need to immediately apply any necessary corrective or preventive actions.”
This is however not a new requirement. PMS was already mandatory under the former Directives (MDD, AIMDD, and IVDD), although less explicitly and extensively described in the legal texts. PMS, or the collection and review of information in post-production phases, are also expected in important international standards like ISO 13485 on QMS for medical devices, ISO 14971 on risk management for medical devices, or IEC 63204 on medical device software lifecycle management.
The PMS system must be established for each device, in consideration to the device type and its risk class, and it must be an integral part of the manufacturer’s Quality Management System (QMS). In brief, PMS is a device-specific process, not just a series of documents.
As depicted in the drawing below, the PMS process involves collecting and analyzing relevant data on the quality, performance and safety of a device, which lead to identifying, implementing and monitoring the necessary preventive and corrective actions. The process must be documented in a PMS Plan and result in a PMS Report or Periodic Safety Update Report (PSUR), depending on the device class. See also chapter “What does PMS entail under the EU MDRand IVDR?”
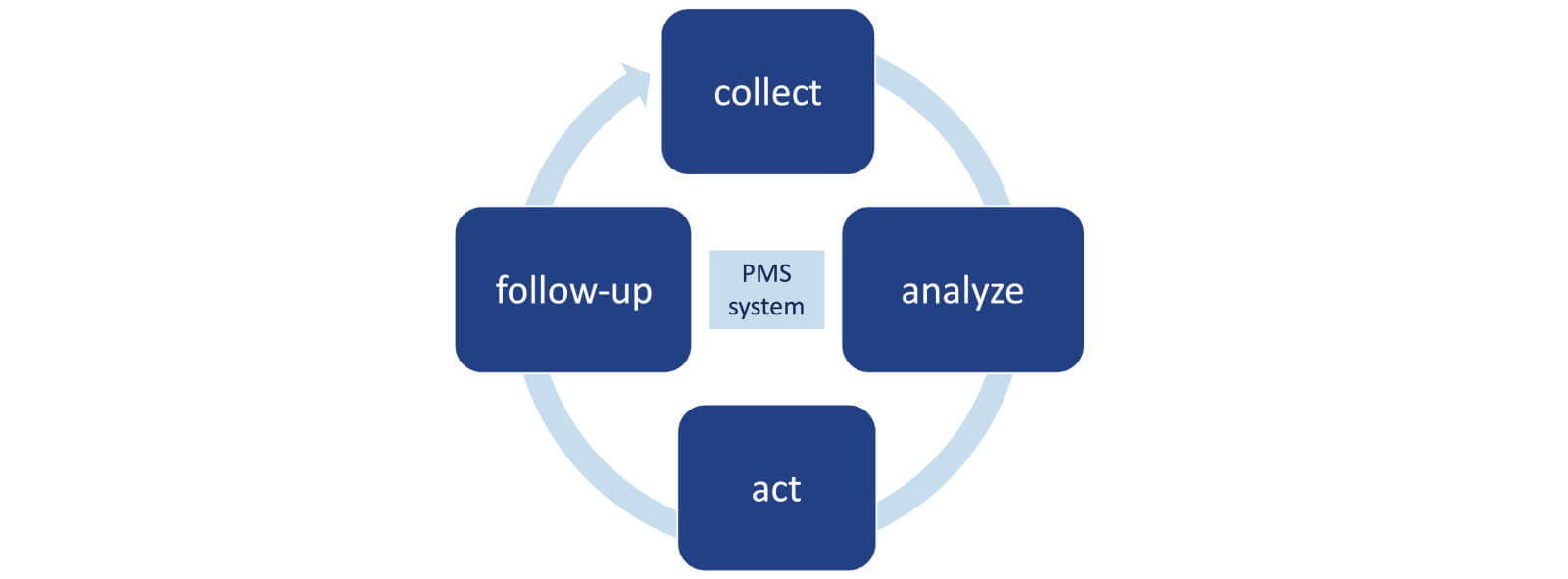
The PMS process is to be continuously run throughout the entire lifetime of the device and is closely intertwined with risk management and clinical/performance evaluation processes.
Is PMS the same as Market Surveillance?
Not at all.
Also described in Chapter VII of the EU MDR and IVDR, market surveillance refers to the compliance controls and enforcement deployed by national competent authorities. It is not the same as the PMS system that the manufacturer must implement.
Market surveillance is conducted by competent authorities via a pre-established plan and may involve:
- announced and unannounced audits of economic operators,
- sampling of devices,
- confiscation of devices that present an unacceptable risk or are falsified.
Competent authorities may request at any time device documentation, information or samples from a manufacturer, its authorized representative or importer/distributor.
The market surveillance plans are established based on risk and Vigilance data or complaints reported to them but this does not mean that low-risk devices with no incident reports are excluded from such inspections. For example, the Swiss competent authority, Swissmedic, conducted an inspection of 27 Class I device manufacturers in March 2023.
What medical devices require PMS activities?
All medical devices (incl. IVDs) placed on the market are subject to the PMS obligations under Chapter VII of the EU MDR or IVDR, as applicable.
This means that “legacy” devices must comply with the PMS requirements in the new Regulations in lieu of the corresponding requirements in the former Directives (MDD, AIMDD, and IVDD), with the following exceptions:
- For legacy non-IVDs, according to guidance document MDCG 2021-25, the output of the PMS data review and analysis does not need to result in the revision of the technical documentation in accordance with Annexes II and III, since legacy devices do not need such technical documentation.
- For legacy IVDs, according to guidance document MDCG 2022-8, a Periodic Safety Update Report (PSUR) which is required for upper risk classes in the IVDR is not expected and the PMS Report suffices, since the former IVDD did not provide for classification of devices in risk classes as it is the case in the IVDR.
In our day-to-day consultancy work, we see that many “legacy” manufacturers have not yet adapted their PMS processes to the EU MDR or IVDR, and that Notified Bodies do not appear to be enforcing this requirement strictly enough. Consequently, PMS Plans and resulting PSURs are out of compliance and make a transition to the EU MDR or IVDR more labor intensive.
Moreover, PMS requirements in the EU MDR also apply to:
- Custom-made devices (CMD), i.e. those tailored to a specific patient upon prescription by a healthcare professional, with the following particularities since for each CMD there is only one model:
- PMS activities are to be applied to groups of similar devices (i.e. same intended purpose, materials, processes used, main design) and not to each individual CMD.
- A communication channel is needed between the manufacturer and the relevant healthcare professionals or concerned patients, to collect the feedback on a given CMD quality, performance and safety.
- For Class III implantable CMDs (which require Notified Body certification of the manufacturer’s QMS), PSURs do not need to be sent to the Notified Body. They just must be part of the CMD documentation that is required under EU MDR Annex XIII.
- Annex XVI products, i.e. products without an intended medical purpose that are governed by the EU MDR, in alignment with the corresponding Common Specifications (CS), Regulation (EU) 2022/2346.
Annex XVI products of class IIa, IIb and III will require a PSUR, once the EU MDR becomes applicable, which in consideration of the amended transitional provisions in the CS (per Regulation (EU) 2023/1194) will be:- 31-Dec-2028 if no clinical trials are planned, or
- 31-Dec-2029 if there are planned or ongoing clinical trials.

In our day-to-day consultancy work, we see that many “legacy” manufacturers have not yet adapted their PMS processes to the EU MDR or IVDR.
Conversely, no separate PMS process is required for Procedure Packs and Systems, as referred to in EU MDR Art. 22 or MDD Art. 12, because these are not additionally CE-marked with respect to the individual devices contained in them. Moreover, assemblers of Procedure Packs and Systems do not necessarily correspond to the legal manufacturers of those individual devices and, thus, do not have access to or maintain the technical documentation.
Who is responsible for PMS?
PMS must be driven by the manufacturer.
However, the activities are to be carried out by the manufacturer in cooperation with other economic operators, since marketed devices may transit in the distribution chain through importers, distributors and resellers, who are also in the position to provide important feedback on the behavior of the device once in use.
Within the manufacturer, the Person Responsible for Regulatory Compliance (PRRC) must oversee PMS activities to ensure that the obligations are complied with. And, whereas PMS obligations under the EU MDR and IVDR apply also to legacy devices, a PRRC is not required for legacy devices, as clearly indicated in both MDCG 2021-25 for non-IVDs and MDCG 2022-8 for IVDs.
What does PMS entail under the EU MDR and IVDR?
Simply put, the PMS process is about collecting, analyzing, acting upon, and following-up on PMS data.
Per EU MDR and IVDR Annex III section 1(a), PMS data must include:
- Vigilance reported cases, i.e. serious incidents and Field Safety Corrective Actions,
- non-serious incidents and any undesirable side-effects,
- information from Vigilance trend reporting,
- other complaints and feedback from users, distributors/importers, that are not Vigilance cases,
- scientific or technical literature,
- information from databases and/or registries, and
- publicly available information about similar devices.
In addition, EU MDR Art. 83(4) and IVDR Art .78(4) introduce a new requirement to inform competent authorities and, where applicable, Notified Bodies, about any corrective or preventive action (CAPA) identified in the course of the PMS activities. These CAPAs become an additional PMS dataset and, according to guidance document MDCG 2022-21, they would correspond to those related to:
- devices already placed on the EU market, i.e. not CAPA opened during development,
- device-related issues that might impact its safety, performance or quality, including also QMS issues with such impact,
- a voluntary suspension of sales, which is not a commercial decision,
- the need to evaluate new/modified benefits and risks identified through PMS activities.
Because the sources of PMS data required are extensive and the review periods mandated by the EU MDR or IVDR are tight, it is important to allocate sufficient resources to this process. PMS activities can be easily outsourced to third parties. See more about “How can Decomplix help”.
Like any other QMS process, PMS must be detailed in a procedure that describes how the activities are planned (PMS Plan), deployed, and documented (PMS Report or PSUR) by the manufacturer. See also chapter “How to prepare a PMS Plan, a PMS Report or a PSUR?”
The outputs of the PMS procedure must ensure alignment with the purposes detailed in EU MDR Art. 83(3) or IVDR Art. 78(3), which include:
- update the benefit-risk determination (and improve risk management)
- update the design and manufacturing information, the instructions for use (IFU) and labelling
- update the clinical/performance evaluation
- update the Summary of Safety (& Clinical) Performance (SSCP/SSP), when applicable
- identify the need for Corrective/Preventive Action (CAPA) or Field Safety Corrective Action (FSCA)
- identify options to improve device usability, performance and safety
- when relevant, contribute to PMS of other devices,
- detect and report Vigilance trends, and
- update the device’s technical documentation accordingly.
As such, the PMS procedure will need links to CAPA, design, manufacturing, risk management, labelling, clinical/performance evaluation, and technical documentation procedures.
An example of PMS procedure’s inputs and outputs is presented below.
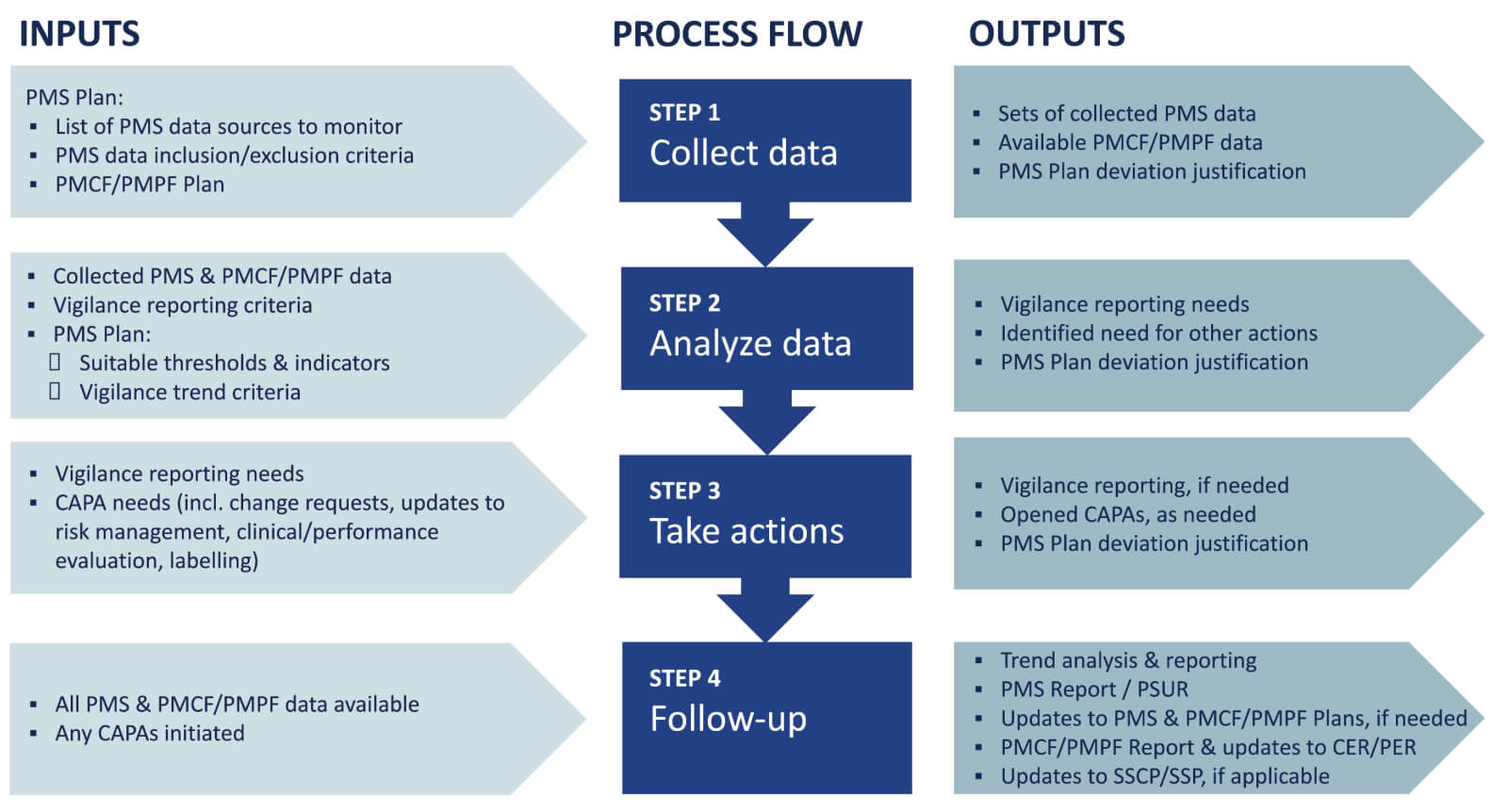
You can also find generic guidance in the technical report ISO/TR 20416 on post-market surveillance for medical device manufacturers. In particular, the annexes provide useful examples of PMS data sources, data analysis methods, and PMS plans.
How to prepare a PMS Plan, a PMS Report or a PSUR?
Let’s start from the end: the Periodic Safety Update Report (PSUR).
A PSUR is always required for:
- Class Ila, Class IIb and Class III medical devices, whether already CE-marked under the EU MDR or still in “legacy” status, i.e. still CE-marked under the MDD/AIMDD.
- Class C and Class D IVDs already CE-marked under the IVDR.
PSUR generation is a recurrent activity, at least annually (for Class IIb/III non-IVDs or Class C/D IVDs) or every 2 years (for Class IIa non-IVDs), until the end of the device lifetime. PSURs must be part of the technical documentation for the concerned device, as specified in Annex III of the EU MDR/IVDR.
Guidance document MDCG 2022-21 is very prescriptive regarding the contents and structure of a PSUR under the EU MDR, and can be extrapolated for the IVDR in the absence of a specific guideline.
Annex I of MDCG 2022-21 presents the template for a PSUR. The picture below shows an example of a table of contents resulting from this template. The expectation of Notified Bodies is that the manufacturer follows the structure as close as possible.

Annex II of MDCG 2022-21 provides the examples for the tabular presentation of PMS data in a PSUR and Annex III explains how the datasets should be presented: per Basic UDI-DI, per region, year to year, and using IMDRF Adverse Event Terminology (AET) terms and codes.
The use of IMDRF AET codes/terms is a challenge that is frequently underestimated by many manufacturers. Most historical data is not yet coded according to these codes, which in addition evolve in time, as IMDRF AET working group periodically reviews them. Manufacturers who have not taken the time to consistently implement the use of IMDRF AET codes in their complaint management and Vigilance systems have a hard time completing a PSUR with PMS data that is comparable from one PMS review period to the next one.

In brief, the PMS Plan is meant to be a “condensed” PMS SOP that applies to a specific device or device group.
In contrast, there is no MDCG guidance document for a PMS Report that is required for lower risk classes and for “legacy” IVDs. The contents of a PMS Report must simply comply with EU MDR Art. 85 or IVDR Art. 80, i.e. a summary of the results and conclusions of the analyses resulting from the PMS Plan together with a rationale and description of any preventive and corrective actions taken. PMS Reports can be updated when necessary. In practice, manufacturers generate PMS Reports mostly based on MDCG 2022-21, particularly when their portfolio includes devices of higher risk classes that require a PSUR.
Both the PSUR and the PMS Report are intended to document the results of the PMS Plan. Per Annex III of the EU MDR and IVDR, the PMS Plan shall cover at least:
- the proactive and systematic process for PMS data collection.
- the effective and appropriate PMS data assessment process and methods.
- suitable indicators and threshold values for continuous reassessment of the benefit-risk analysis and risk management.
- the effective and appropriate methods and tools for complaint investigation and market feedback analysis.
- the methods and protocols for trend analysis (i.e. the detection of statistically significant increase in frequency/severity of incidents).
- the methods and protocols for effective communication with competent authorities, notified bodies, economic operators and users.
- a reference to the manufacturer’s Vigilance reporting procedures.
- systematic procedures to identify and initiate appropriate measures, incl. CAPA.
- tools to trace and identify devices subject to CAPA.
- the Post-market Clinical Follow-up (PMCF) or Post-market Performance Follow-up (PMPF) Plan, or a justification as to why a PMCF/PMPF is not applicable.
In brief, the PMS Plan is meant to be a “condensed” PMS SOP that applies to a specific device or device group.
Although the list above is extremely explicit and only mentions “a reference” once, with respect to Vigilance procedures, most manufacturers still complete PMS Plans under the EU MDR or IVDR like in the old days, with a simple table of references to QMS procedures and work instructions, no details about indicators and thresholds to trigger action (or mistaking Vigilance trend analysis for such thresholds and indicators) and an invalid justification as to why PMCF/PMPF is not applicable.
The easiest and most compliant manner of preparing a PMS Plan is through “reverse engineering” from MDCG 2022-21: every element that must end up in a PSUR should be part of the corresponding PMS Plan.
What are “suitable thresholds and indicators”?
It is a common misconception to consider that the PMS thresholds and indicators are the same as the triggers for Vigilance trend reporting. Although there is some overlap, they are not exactly the same, and are in fact mentioned in different subparagraphs of Annex III section 1(b) of the EU MDR and IVDR.
The scope of Vigilance trend detection is limited to non-serious incidents to be able to identify trends that must be reported, per EU MDR Art. 88 or IVDR Art. 83, which are those with significant impact on the benefit-risk profile of the device and have led or may lead to unacceptable risks. In the case of non-IVDs, this applies also to expected undesirable side-effects, and in the case of IVDs, to expected erroneous results. The manufacturer must establish the significant increase by comparing the data within a specific time period (usually the PMS review period) with the foreseeable frequency or severity of such incidents. The methodology for determining such trends, which is expected to be part of an upcoming MDCG guidance document, must be described in the PMS Plan. In the meantime, Appendix C in the old guideline from the Global Harmonization Task Force, GHTF/SF2/N36R7, proves very helpful.
The scope of PMS indicators and threshold values that must be described in the PMS Plan is broader since these are needed for all datasets, in order to continuously reassess the benefit-risk profile and to identify any actions that might need to be taken. The indicators and thresholds should be meaningful for the type of PMS dataset analyzed and must compare the present PMS review period with the previous ones for a total 4-year timeframe, in accordance with MDCG 2022-21. The methods can be quantitative (for example, the statistical analysis needed to identify Vigilance trend), qualitative (for example, detection of shifts in the state-of-the-art or new risks in the scientific literature reviewed), or semi-quantitative(for example, the meta-analysis of published studies, by mixing both qualitative sorting and quantitative analysis). The technical report ISO/TR 20416 provides some guidance for the selection of appropriate methods.
What are the differences between PMS and PMCF/PMPF?
Post-market CLINICAL Follow-up (PMCF) under the EU MDR and Post-market PERFORMANCE Follow-up (PMPF) under the IVDR refer to the process required to continuously update the Clinical Evaluation (for non-IVDs) and the Performance Evaluation (for IVDs), respectively.
Both the EU MDR (in Annex XIV, Part B, point 5) and IVDR (in Annex XIII, Part B, point 4) require that the PMCF/PMPF be “addressed in the manufacturer’s post-market surveillance plan”. In brief, the PMCF or PMPF Plan must be part of the corresponding PMS Plan and a summary of the PMCF/PMPF report is expected to be integrated in the PSUR.
Some of the PMCF/PMPF data, what is called “general” PMCF/PMPF, overlaps with PMS data, as shown in the picture below.
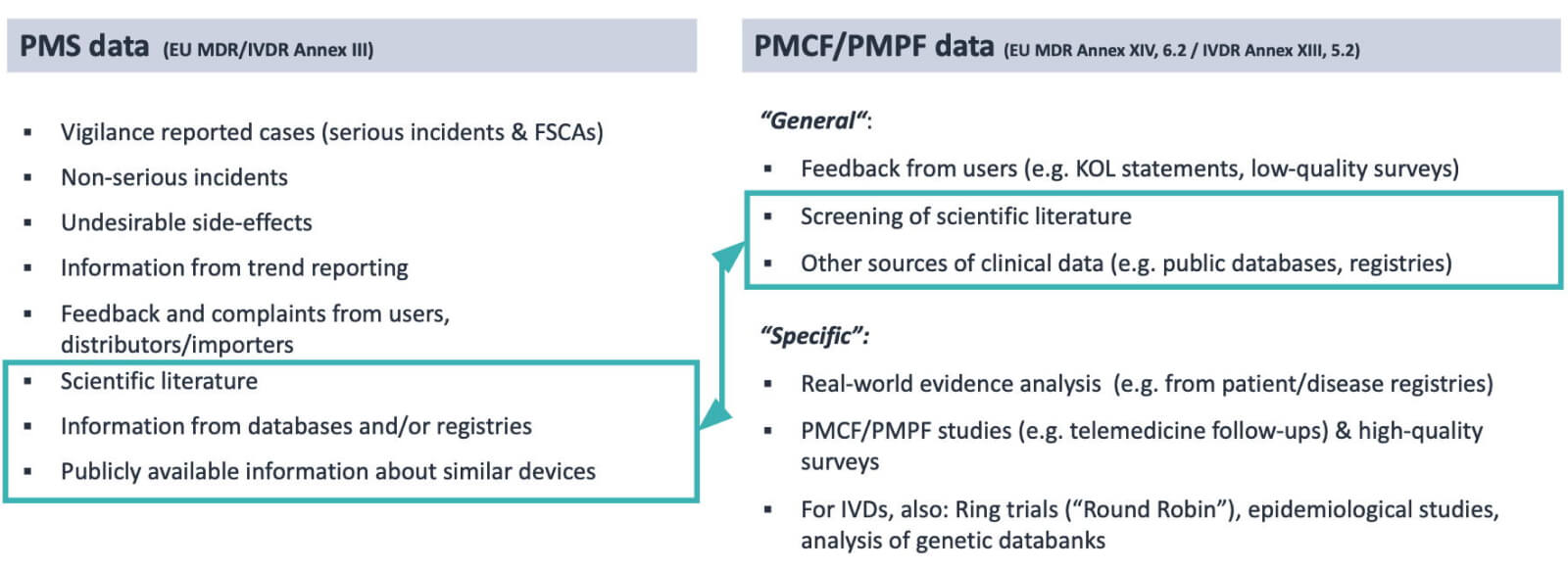
Because PMCF/PMPF is the proactive collection of any clinical/performance data to keep the Clinical Evaluation Report (CER) or Performance Evaluation Report (PER) up-to-date, it makes sense to report the common, overlapping data as the PMCF/PMPF data rather than PMS data.
How Decomplix can help
Decomplix offers consultancy services in all regulatory and quality assurance matters relative to medical devices. We can support your company in fulfilling all PMS and PMCF/PMPF requirements, and ensuring compliance in the most efficient manner. If you wish to discuss your needs with us, please contact us for a non-binding CE marking quote.
Further reading
- Certification of a Quality Management System according to ISO 13485
- Risk Management for Medical Devices under EU MDR and ISO 14971
- Medical device software (MDSW) under EU MDR and IVDR
- “Legacy” medical devices and IVDs under EU legislation
- Annex XVI and EU MDR – Guide to regulation of products without intended medical purpose



