
Schweizer Importeure von Medizinprodukten – rechtliche Anforderungen
Sind Sie im Unklaren über die Anforderungen an den Import von Medizinprodukten oder IVD in die Schweiz? Haben Sie vom “virtuellen” Schweizer Importeur gehört und fragen Sie sich, ob der Ihnen nützlich sein könnte? Verkaufen Sie Medizinprodukte, IVD oder medizinisches Zubehör in der Schweiz und fragen sich, ob Sie als Schweizer Importeur gelten? Hier finden Sie alles, was Sie darüber wissen müssen.
Ersetzt die Version vom 27.6.2022
Wichtige Erkenntnisse:
- Jedes in der Schweiz ansässige Unternehmen, das Medizinprodukte oder In-vitro-Diagnostika vom Ausland auf den Schweizer Markt bringt, wird zum Schweizer Importeur.
- Das Konzept des “Inverkehrbringens” ist bedeutend. Damit werden auch Apotheken, Optiker, Supermärkte, Online-Shops oder andere Abgabestellen zu Schweizer Importeuren.
- Es gibt keinen “virtuellen” Medizinprodukteimporteur. Seien Sie vorsichtig bei der Wahl Ihres Importeurs.
- Es ist wichtig, dass Sie sich mit Ihren Compliance-Pflichten als Schweizer Importeur vertraut machen. Wenn Sie glauben, dass Sie fachliche Unterstützung benötigen, kontaktieren Sie uns bitte.
Inhalt:
Warum wird für Medizinprodukte, einschliesslich In-vitro-Diagnostika (IVD), plötzlich ein Schweizer Importeur benötigt?
Die Einfuhr von CE-gekennzeichneten Medizinprodukten und IVD in die Schweiz wurde früher nicht von der Einfuhr in andere EWR-Länder unterschieden, da die europäischen Richtlinien 93/42/EWG über Medizinprodukte (MDD), 90/385/EWG über aktive implantierbare Medizinprodukte (AIMDD) und 98/79/EG über In-vitro-Diagnostika (IVDD) im Rahmen der alten Schweizer Medizinprodukteverordnung (MepV, SR 812.213), die auf dem Abkommen über die gegenseitige Anerkennung (MRA) zwischen der Schweiz und der EU beruht, vollständig übernommen wurden. Dies ist nun nicht mehr der Fall.
Seit dem 26. Mai 2021, dem Datum des Inkrafttretens der neuen Verordnung (EU) 2017/745 über Medizinprodukte (EU-MDR), gilt die Schweiz als Drittland in Bezug auf die Auswirkungen der EU-MDR. Zum gleichen Zeitpunkt hat die Schweiz eine revidierte Fassung ihrer Medizinprodukteverordnung (MepV, SR 812.213) in Kraft gesetzt, die aufgrund des Fehlens eines aktualisierten MRA schweizerische Anforderungen einführt. Diese Anforderungen haben zum Teil Auswirkungen auf in der Schweiz ansässige Unternehmen, die Medizinprodukte importieren. Dasselbe geschah anschliessend für IVD durch die neue Schweizer Verordnung über In-vitro-Diagnostika (IvDV), die am 26. Mai 2022 in Kraft trat.
«Die Schweiz gilt als Drittland in Bezug auf die Auswirkungen der EU-MDR.»
Um die Situation rückgängig zu machen, müsste das MRA aktualisiert werden, was wahrscheinlich in nächster Zeit nicht geschehen wird, da die EU-Kommission das MRA nicht verhandeln oder aktualisieren wird, solange keine Einigung zwischen beiden Parteien erzielt wurde. Die Entscheidung der Schweiz, das institutionelle Abkommen (InstA) nicht zu unterzeichnen und den Prozess einseitig zu stoppen, hat weitreichende und langfristige Konsequenzen. Die aktuelle politische Situation in der Schweiz macht eine Lösung in absehbarer Zeit sehr unwahrscheinlich.
Ohne ein aktualisiertes MRA wird die Schweiz weiterhin als “Drittland” von den Auswirkungen der MDR und IVDR der EU betroffen sein, und die EWR-Länder werden im Rahmen der schweizerischen Gesetzgebung über Medizinprodukte und IVDs weiterhin als “Drittländer” gelten.
Was ist ein Importeur von Medizinprodukten und wer kann diese Rolle in der Schweiz übernehmen?
Ein Schweizer Medizinprodukteimporteur ist wie folgt definiert (Art. 4 Abs. 1 Bst. h MepV und Art. 4 Abs. 1 Bst. g IvDV):
“jede in der Schweiz niedergelassene natürliche oder juristische Person, die ein Produkt aus dem Ausland auf dem Schweizer Markt in Verkehr bringt”
Das bedeutet, dass jedes Unternehmen mit Sitz in der Schweiz, das Produkte aus dem Ausland auf den Schweizer Markt bringt, zum Importeur wird. Mit anderen Worten: Ein Unternehmen wird “durch Tätigkeit” zum Schweizer Importeur und nicht durch die Benennung durch den ausländischen Hersteller.
In der Praxis schränkt dies die Möglichkeit eines ausländischen Herstellers ein, Produkte direkt an Vertriebsunternehmen oder Einzelhändler zu liefern, wenn diese Unternehmen nicht bereit sind, die Pflichten eines Schweizer Importeurs zu übernehmen. Auch Apotheken, Supermärkte, Online-Shops und andere Abgabestellen, die direkt aus dem Ausland bezogene Produkte verkaufen, gelten als Schweizer Importeure.
Es ist zu beachten, dass “Inverkehrbringen” die erstmalige Bereitstellung auf dem Schweizer Markt bedeutet (Art. 4 Abs. 1 Bst. b MepV/IvDV). “Bereitstellung” wiederum ist definiert als jede Abgabe eines Produkts (ausser einem Prüfprodukt) zum Vertrieb, Verbrauch oder zur Verwendung innerhalb der Schweiz und nicht das Über-die-Grenze-bringen, was fälschlicherweise oft angenommen wird.
Swissmedic führt den Begriff der Bereitstellung in Kapitel 3 ihres Merkblatts Pflichten Wirtschaftsakteure wie folgt weiter aus:
“Die Bereitstellung eines Produkts setzt ein Angebot oder eine (schriftliche oder mündliche) Vereinbarung zwischen zwei oder mehr juristischen oder natürlichen Personen in Bezug auf die Übertragung des Eigentums, des Besitzes oder sonstiger Rechte hinsichtlich des betreffenden Produkts nach dessen Herstellung voraus, was nicht zwingend die physische Übergabe des Produkts erfordert. Diese Übertragung kann entgeltlich oder unentgeltlich erfolgen.”
Wird eine solche “Übertragung des Eigentums, des Besitzes oder sonstiger Rechte” von einem bestimmten Schweizer Unternehmen vorgenommen, so ist dieses Unternehmen der Schweizer Importeur.

Die Rolle des Schweizer Importeurs wird bestimmt “durch Tätigkeit”. Es gibt keinen “virtuellen” oder “designierten” Schweizer Importeur.
Beachten Sie, dass “Parallelimporte” (d.h. Lieferungen durch ausländische Unternehmen, die nicht Hersteller sind) aus regulatorischer Sicht nicht anders behandelt werden als Direktimporte vom ausländischen Hersteller. In beiden Fällen wäre das Unternehmen mit Sitz in der Schweiz, das die Produkte auf den Schweizer Markt bringt, ein Schweizer Importeur.
Was sind die Pflichten des Schweizer Importeurs? Können diese delegiert werden?
Die Pflichten eines Schweizer Importeurs gemäss MepV/IvDV sind relativ einfach, da sie denen eines Importeurs gemäss EU-MDR/IVDR Art. 13 entsprechen. Sie lassen sich in folgende Tätigkeitsbereiche unterteilen (Art. 53 MepV und Art. 46 IvDV):
Überprüfung der Konformität
Vor dem “Inverkehrbringen” eines Produkts in der Schweiz muss der Schweizer Importeur überprüfen, ob es den Anforderungen entspricht (Art. 53 Abs. 1 MepV und Art. 46 Abs. 1 IvDV), d.h.:
- ob das Produkt die CE-Kennzeichnung trägt,
- ob die Konformitätserklärung ausgestellt wurde,
- ob der Hersteller einen Schweizer Bevollmächtigten benannt hat,
- ob das Produkt entsprechend der MepV/IvDV gekennzeichnet ist und
- ob gegebenenfalls eine UDI vom Hersteller zugewiesen wurde.
Konformitätserklärungen und gegebenenfalls Zertifikate der Benannten Stelle sind vom Schweizer Importeur während 10 Jahren (15 Jahre bei implantierbaren Produkten) nach dem letzten Inverkehrbringen aufzubewahren (Art. 13 Abs. 9 EU-MDR/IVDR).
Darüber hinaus wird vom Schweizer Importeur erwartet, dass er nachweisen kann, dass die Konformitätsbewertung durchgeführt wurde und dass das Produkt konform ist (Art. 21 Abs. 2 MepV und Art. 17 Abs. 2 IvDV). Dies bedeutet einen erheblichen Aufwand für jeden Schweizer Importeur, insbesondere bei der Überprüfung der verlängerten Gültigkeit von EG-Zertifikaten für “Legacy”-Produkte unter der MDD/AIMDD. Swissmedic erwartet von Schweizer Importeuren, dass sie die Plausibilität überprüfen:
- der Selbstdeklaration des Herstellers gemäss EU-MDR Art. 120 Abs. 3 Bst. c
- des Bestätigungsschreibens der Benannten Stelle, dass der Hersteller ein Gesuch um EU-MDR-Zertifizierung gestellt hat.
Schweizer Importeure dürfen ein Produkt nicht in Verkehr bringen, wenn sie Grund zur Annahme haben, dass es nicht konform ist (Art. 53 Abs. 3 MepV und Art. 46 Abs. 3 IvDV).
Im Falle einer Nichtkonformität muss ein Verfahren vorhanden sein, das vorsieht, den Hersteller und seinen Schweizer Bevollmächtigten zu informieren.
Geeignete Lager- und Transportbedingungen
Solange ein Produkt unter der Verantwortung des Schweizer Importeurs steht, müssen die Lagerungs-/Transportbedingungen den allgemeinen Sicherheits- und Leistungsanforderungen entsprechen (Art. 13 Abs. 5 EU-MDR/IVDR), bzw. den grundlegenden Anforderungen für “Legacy-Produkte”, die noch unter die MDD/AIMDD/IVDD fallen. Darüber hinaus muss der Schweizer Importeur, sofern vorhanden, die vom Hersteller festgelegten Lager- und Transportbedingungen einhalten.
Rückverfolgbarkeit der Produkte
Der Schweizer Importeur muss mit dem Hersteller oder dessen Schweizer Bevollmächtigten zusammenarbeiten, um eine Rückverfolgbarkeit der Produkte sicherzustellen (Art. 64 MepV und Art. 57 IvDV). Dazu gehört, dass er Swissmedic auf Anfrage die Identität der Wirtschaftsakteure mitteilt, von denen er ein Medizinprodukt erworben hat, wie auch die der Wirtschaftsakteure oder Kunden, an die er ein Medizinprodukt abgegeben hat (Art. 47 Bst. c des Schweizerischen Heilmittelgesetzes). Diese Meldepflicht gilt während mindestens 10 Jahren (15 Jahre bei implantierbaren Produkten) ab dem Zeitpunkt des Erwerbs oder der Lieferung.
Register für Beschwerden, nicht konforme Produkte, Rückrufe und Rücknahmen
Der Schweizer Importeur muss ein Register der Beschwerden, der nicht konformen Produkte sowie der Rückrufe und Rücknahmen führen und dem Hersteller, seinem Schweizer Bevollmächtigten und den Händlern alle von ihnen angeforderten Informationen zur Verfügung stellen, damit diese den Beschwerden nachgehen können (analog zu Art. 13 Abs. 6 EU-MDR/IVDR).
Kommunikation
Der Schweizer Importeur leitet alle Beschwerden und Vorkommnisse unverzüglich an den Hersteller und seinen Schweizer Bevollmächtigten weiter (analog zu Art. 13 Abs. 8 EU-MDR/IVDR). Dies gilt auch im Fall von Parallelimporten, wo der Schweizer Importeur keine direkte wirtschaftliche Verbindung zum Hersteller hat. Stellt ein nicht konformes Produkt ein ernsthaftes Risiko dar, informiert der Schweizer Importeur unverzüglich Swissmedic, insbesondere unter Angabe der Nichtkonformität und der ergriffenen Korrekturmassnahmen (analog zu Art. 13 Abs. 7 EU-MDR/IVDR).
Zusammenarbeit
Im Falle von Korrekturmassnahmen zur Herstellung der Konformität eines Produkts (inkl. Rückrufe und Rücknahmen) kooperiert der Schweizer Importeur mit Swissmedic, falls vorhanden mit der Benannten Stelle sowie dem Hersteller und seinem Schweizer Bevollmächtigten (analog zu Art. 13 Abs. 10 EU-MDR/IVDR).
Registrierung bei Swissmedic (CHRN)
Der Schweizer Importeur muss sich bei Swissmedic registrieren lassen, was den Erhalt der Einmaligen schweizerischen Registrierungsnummer (CHRN) beinhaltet (Art. 55 Abs. 1 MepV und Art. 48 Abs. 1 IvDV). Für diese Registrierung ist der Zeitpunkt des ersten Inverkehrbringens entscheidend:
- Wenn der Importeur entweder ein Medizinprodukt nach dem 26. Mai 2021 oder ein IVD nach dem 26. Mai 2022 zum ersten Mal auf den Schweizer Markt bringt, muss die Registrierung innerhalb von drei Monaten erfolgen.
- Hat der Importeur vor dem 26. Mai 2021 erstmals EU-MDR-konforme Produkte in der Schweiz in Verkehr gebracht, so hat er die Registrierung vor dem 26. November 2021 vornehmen müssen.
- Hat der Importeur IVDR-konforme Produkte vor dem 26. Mai 2022 erstmals in der Schweiz in Verkehr gebracht, so hat er die Registrierung vor dem 26. November 2022 vornehmen müssen.
- Hat der Importeur vor dem 26. Mai 2021 “Legacy”-Medizinprodukte oder vor dem 26. Mai 2022 “Legacy”-IVD erstmals auf den Schweizer Markt gebracht, so besteht keine Registrierungspflicht.
Beachten Sie, dass für Schweizer Importeure, die sowohl IVD als auch Medizinprodukte handeln, die Registrierung bei Swissmedic nur einmal erforderlich ist. Nachträgliche Änderungen der gemeldeten Angaben des Schweizer Importeurs (z.B. Adressänderung) sind innerhalb einer Woche Swissmedic zu melden (Art. 55 Abs. 2 MepV und Art. 48 Abs. 2 IvDV).
Angabe der Daten des Schweizer Importeurs
Die Angaben des Schweizer Importeurs müssen auf dem Produkt, seiner Verpackung oder einem Begleitdokument (z.B. Rechnung, Lieferschein, Garantie oder ähnliches) erscheinen (Art. 53 Abs. 2 MepV und Art. 46 Abs. 2 IvDV). Siehe auch: Müssen ausländische Hersteller die Kennzeichnung ändern, um den Schweizer Importeur anzugeben?
Alle oben genannten Pflichten müssen offiziell vom Schweizer Importeur übernommen werden, d.h. der Schweizer Importeur bleibt für alle seine Verpflichtungen verantwortlich und rechenschaftspflichtig gegenüber Swissmedic. Allerdings können gewisse operative Tätigkeiten, unter Kontrolle des Schweizer Importeurs, an Dritte delegiert werden. So könnten beispielsweise die Lagerung an einen in der Schweiz ansässigen Logistikdienstleister oder regulatorische Aufgaben (z.B. die “Fern”-Überprüfung eingehender Waren) an eine andere Einheit innerhalb eines grossen Unternehmens oder an ein Beratungsunternehmen für regulatorische Fragen delegiert werden.

Jede Delegation von Tätigkeiten muss in einer Qualitätsvereinbarung zwischen den beteiligten Parteien dokumentiert werden.
Die Delegation von regulatorischen Aufgaben bedeutet nicht, dass das Unternehmen, das diese Tätigkeiten durchführt, als Schweizer Importeur angesehen werden kann. Frage 16 des Leitfadens MDCG 2021-27 über die Rolle von Importeuren und Händlern gemäss EU-MDR/IVDR untersagt die Delegation der rechtlichen Verantwortung von einem Importeur an einen anderen ausdrücklich, da Importeure eine wesentliche Aufgabe bei der Überwachung der Lieferkette und der Gewährleistung der Rückverfolgbarkeit haben.
Jede Delegation von Tätigkeiten muss in einer Qualitätsvereinbarung zwischen den beteiligten Parteien dokumentiert werden.
Kann ich als ausländischer Medizinproduktehersteller einen einzigen Schweizer Importeur benennen, der die entsprechenden Pflichten “virtuell” übernimmt?
Wie bereits erwähnt, wird ein Schweizer Unternehmen durch seine Tätigkeit zum Schweizer Importeur. Es gibt also nur eine kommerzielle, aber keine regulatorische Benennung von Schweizer Importeuren.
Ein paar Beispiele:
- Ein regulatorisches Beratungsunternehmen bietet Dienstleistungen als “virtueller” Schweizer Importeur für ausländische Hersteller an. Das Unternehmen übernimmt einige oder alle regulatorischen Tätigkeiten, z.B. das Führen des Registers für Beanstandungen, Nonkonformitäten und Rückrufe/Rücknahmen, oder die Kommunikation mit Swissmedic oder die Fernüberprüfung der Konformität für jedes in Verkehr gebrachte Produkt im Namen des Unternehmens, das dieses Produkt tatsächlich importiert und auf dem Schweizer Markt in Verkehr bringt.
In diesem Fall kann das Beratungsunternehmen nicht als Schweizer Importeur angesehen werden und sollte sich nicht bei Swissmedic als Schweizer Importeur registrieren. Diesem Dienstleister werden lediglich einige operative Aufgaben “delegiert”.
- Ein ausländischer Hersteller “benennt” seine Schweizer Tochtergesellschaft als Schweizer Importeur, liefert die Produkte aber direkt an verschiedene in der Schweiz ansässige Händler. Zwei Fälle sind möglich:
- Wenn der “Eigentumsübergang” der in die Schweiz gebrachten Produkte zwischen dem ausländischen Hersteller und seiner Tochtergesellschaft und dann von der Tochtergesellschaft an den Schweizer Vertriebshändler oder Einzelhändler erfolgt, kann die Tochtergesellschaft als Schweizer Importeur angesehen werden, auch wenn die Produkte nie an sie versandt oder von ihr gelagert werden.
- Wenn der “Eigentumsübergang” der in die Schweiz gebrachten Produkte nur zwischen dem ausländischen Hersteller und dem Schweizer Grossverteiler oder Einzelhändler stattfindet, kann die Tochtergesellschaft nicht als Schweizer Importeur angesehen werden. Stattdessen wird der Schweizer Grossverteiler oder Einzelhändler zum Schweizer Importeur.
- Ein ausländischer Hersteller “benennt” ein Schweizer Unternehmen als seinen Alleinimporteur, liefert und verkauft die Produkte dann aber direkt an die Krankenhäuser. Zwei Fälle sind möglich:
- Der direkte Versand von ausländischen Herstellern an Spitäler, die die Endverbraucher sind, wird nicht als “Inverkehrbringen” betrachtet, so dass kein Schweizer Importeur erforderlich ist (ebenso ist kein Schweizer Bevollmächtiger). Wenn das Spital die Produkte für den Eigengebrauch importiert, wird es daher nicht als Schweizer Importeur betrachtet.
- Wenn das Spital die Produkte weiterverkauft (oder anderweitig das Eigentum daran überträgt), wird es zu einem Schweizer Importeur.
Ist in jedem Fall ein Schweizer Importeur erforderlich? Was ist mit Mediziproduktesoftware?
Ein Schweizer Importeur ist nicht involviert, wenn es sich um Geschäfte mit Produkten handelt, die nicht in der Schweiz “in Verkehr gebracht” werden (wie in Art. 4 Abs.1 Bst. b MepV/IvDV definiert). Dies ist der Fall für:
- Prüfprodukte.
- Produkte, die direkt aus dem Ausland zum Endverbraucher importiert werden, sei es eine Gesundheitsfachperson, eine Gesundheitseinrichtung oder ein Patient. Dies wird in der Schweiz als direkte Anwendung betrachtet. Im Falle der Gesundheitsfachperson trägt diese die Verantwortung für die Konformität des Produkts. Weitere Details sind im Merkblatt von Swissmedic zur Beschaffung von Medizinprodukten in Gesundheitseinrichtungen zu finden.
- Produkte, die bereits vor Inkrafttreten der MepV/IvDV vom Schweizer Importeur an den Händler übergeben wurden und sich noch im Lager des Händlers befinden, gelten als in der Schweiz nach der alten Gesetzgebung in Verkehr gebracht. Der Schweizer Importeur muss daher nicht rückwirkend gemeldet werden.
Bezüglich Medizinproduktesoftware unterscheiden weder die MepV/IvDV noch die EU-MDR/IVDR zwischen Soft- und Hardware, was die Anforderungen an die Importeure betrifft.
Offensichtlich müssten die Pflichten zu Lager- und Transportbedingungen als Pflichten zur Bereitstellung von Software verstanden werden, da es kein physisches Produkt gibt. Alle anderen Pflichten gelten jedoch unverändert. Ein Schweizer Importeur wäre demnach jedes in der Schweiz ansässige Unternehmen, das Medizinproduktesoftware in der Schweiz in Verkehr bringt, z.B. indem es sie den Endnutzern über seine eigene Plattform zur Verfügung stellt.
Kann der Schweizer Bevollmächtigte auch der Schweizer Importeur sein?
Der Schweizer Bevollmächtigte und der Schweizer Importeur sind unterschiedliche Wirtschaftsakteure mit unterschiedlichen regulatorischen Pflichten im Rahmen der MepV und der IvDV. Die beiden Funktionen werden in der Regel von unterschiedlichen Arten von Unternehmen wahrgenommen, wie weiter unten erläutert wird.
Beide Funktionen können jedoch unter demselben Unternehmen zusammengefasst werden. Ein Unternehmen, das sowohl als Schweizer Bevollmächtigter als auch als Schweizer Importeur auftritt, muss sich bei Swissmedic separat für beide Funktionen als Wirtschaftsakteur registrieren lassen (Art. 55 MepV und Art. 48 IvDV) und erhält von Swissmedic zwei separate Einmalige Schweizer Registrierungsnummern (CHRN).

Die Rolle des Schweizer Importeurs wird bestimmt “durch Tätigkeit”.
Auch wenn die Aufgaben des Schweizer Importeurs die Überprüfung der Einhaltung der Vorschriften und die Meldepflichten umfassen, die mit denen des Schweizer Bevollmächtigten vergleichbar sind, erstrecken sich die Pflichten des Schweizer Importeurs auch auf Lager- und Transporttätigkeiten, einschliesslich des erforderlichen Qualitätsmanagementsystems.
Ein Schweizer Importeur könnte die zusätzliche Rolle des Schweizer Bevollmächtigten übernehmen, sofern er über ausreichende interne regulatorische Ressourcen verfügt. Hingegen ist selten der Fall, dass ein Schweizer Bevollmächtigter die Rolle des Importeurs übernimmt, da die Einfuhr neben der Einhaltung der MepV/IvDV auch zollrechtliche, logistische und steuerliche Tätigkeiten mit sich bringt und Unternehmen, die Dienstleistungen eines Schweizer Bevollmächtigten anbieten, in der Regel regulatorische Beratungsunternehmen ohne Kompetenz in diesen Bereichen sind. Die Rolle eines Schweizer Importeurs ist für einen bestehenden Schweizer Händler natürlich einfacher.
Lesen Sie mehr über die Rolle des Schweizer Bevollmächtigten.
Kann ein Unternehmen gleichzeitig als Schweizer Händler und Schweizer Importeur auftreten?
Nein.
Der Schweizer Importeur und der Schweizer Händler sind gemäss MepV und IvDV separate regulatorische Rollen.
Diese Rollen sind nicht zu verwechseln mit dem Konzept des Vertriebs von Waren im Sinne von Versand oder Verkauf. Natürlich können sowohl Importeure als auch Händler Produkte versenden oder verkaufen, aber diese Aktivitäten sind nicht der entscheidende Faktor.
Der Schweizer Importeur holt Medizinprodukte aus dem Ausland (entweder vom ausländischen Hersteller oder einem ausländischen Händler) und bringt sie auf den Schweizer Markt, d.h. die Weitergabe oder Lieferung der Produkte vom Schweizer Importeur an einen Schweizer Händler, eine Gesundheitseinrichtung oder den Endverbraucher. Dies entspricht dem roten Pfeil in der untenstehenden Infografik von Swissmedic.
Der Schweizer Händler stellt die Produkte nur auf dem Schweizer Markt zur Verfügung, d.h. er liefert sie an andere Schweizer Gross-, Einzelhändler, an eine Gesundheitseinrichtung oder den Endverbraucher weiter. Dies entspricht entweder dem transparenten oder dem grünen Pfeil in der untenstehenden Infografik von Swissmedic.
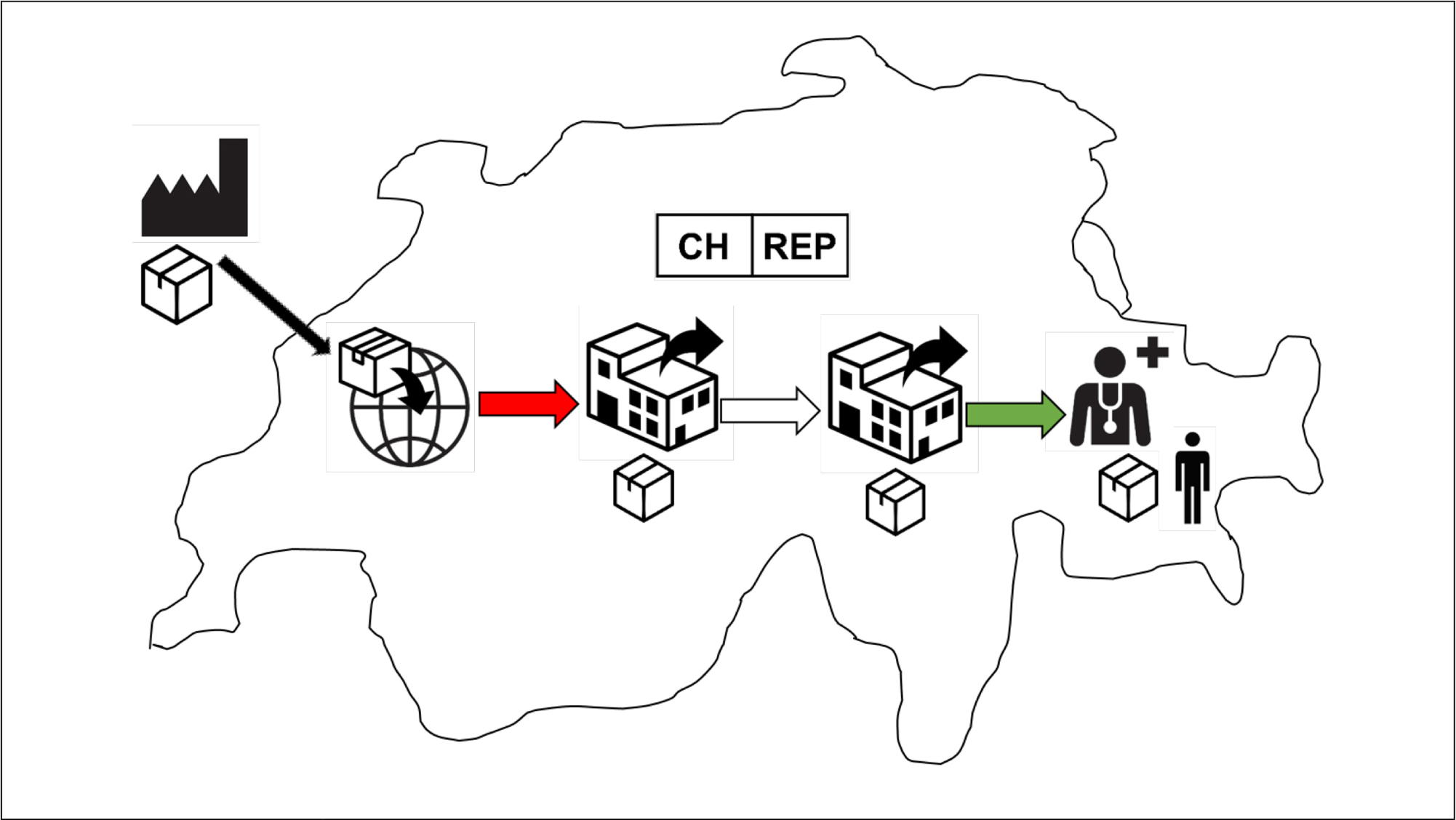
Der Schweizer Importeur und der Schweizer Händler sind gemäss MepV und IvDV separate regulatorische Rollen.
Müssen ausländische Hersteller die Kennzeichnung ändern, um den Schweizer Importeur anzugeben?
Name und Anschrift des Schweizer Importeurs müssen auf dem Produkt, auf der Verpackung oder auf einem Begleitdokument des Produkts angegeben werden (Art. 53 Abs. 2 MepV und Art. 46 Abs. 2 IvDV). Beispiele für Begleitdokumente eines Produkts: Lieferschein, Garantieschein, Zolldokumente, Rechnung, Aufkleber auf der Verpackung oder Gebrauchsanweisung.
Swissmedic gibt im Kapitel 6 ihres Merkblatts zu den Pflichten der Wirtschaftsakteure zusätzliche Informationen zur obligatorischen Angabe des Schweizer Importeurs. Gemäss dem Merkblatt kann ein Begleitdokument dem Produkt beigefügt oder von ihm getrennt sein. Es muss nicht zwingend den Endverbraucher erreichen, der Schweizer Importeur muss jedoch entlang der Lieferkette eindeutig identifizierbar sein, auch wenn es keinen Vertreiber gibt.
Zweck der Informationen ist eine schnelle und eindeutige Identifizierung der Wirtschaftsakteure, die für die betreffenden Produkte verantwortlich sind, z.B. für die Durchführung von Produktrückrufen oder die Meldung von Vorkommnissen.
«Der Schweizer Importeur muss entlang der Lieferkette eindeutig identifizierbar sein.»
In Ermangelung einer klaren Anleitung von Swissmedic empfiehlt Decomplix die Verwendung des Begriffs “CH IMPORTEUR” – in Englisch, Französisch und Italienisch entsprechend: “CH IMPORTER / IMPORTATEUR / IMPORTATORE”. Die Verwendung des Importeursymbols der ISO 15223-1 empfehlen wir zu vermeiden, da es mit dem EU-Importeur verwechselt werden könnte.
Die Etiketten müssen auf Deutsch, Französisch und Italienisch übersetzt werden, siehe: Sprachliche Anforderungen der EU-MDR (Art. 16 Abs. 2 der MepV und Art. 15 Abs. 2 der IvDV). Decomplix ist der Ansicht, dass es nicht notwendig ist, die Stadt der Adresse des Schweizer Importeurs in andere Schweizer Sprachen zu übersetzen, da die Adressen in der Schweiz durch die Postleitzahl bestimmt werden.
Ist das Umetikettieren und Umverpacken durch Schweizer Importeure erlaubt?
Die Anforderungen an Stellen, die Produkte im Rahmen der MepV/IvDV umetikettieren und/oder umpacken, sind dieselben wie die der EU MDR/IVDR.
Dabei sind folgende verschiedene Situationen möglich:
Der Schweizer Importeur übernimmt das Umetikettieren oder Umverpacken als Unterauftragnehmer im Auftrag und unter der Kontrolle des rechtmässigen Herstellers.
In Kapitel 7 des Swissmedic-Infoblattes zu den Pflichten der Wirtschaftsakteure wird klargestellt, dass sich Swissmedic bei der Auslegung der Anforderungen an die Umetikettierung und das Umverpacken auf die europäische Praxis und insbesondere auf die von der Medical Device Coordination Group (MDCG) herausgegebenen Leitfäden stützt. Dies bedeutet, dass die MDCG 2021-26 (Q&A on repackaging and relabelling activities) berücksichtigt werden muss. Abschnitt 2 dieses Leitliniendokuments besagt Folgendes:
“Article 16(2), (3) and (4) of the Regulations do not apply to operators subcontracted by the manufacturer (that may also qualify as importers or distributors), who also carry out relabelling and/or repackaging activities on behalf and under the control of the manufacturer.”
In einem solchen Fall wird erwartet, dass zwischen dem Hersteller und dem Schweizer Importeur eine spezifische Qualitätsvereinbarung für die Tätigkeiten im Unterauftrag abgeschlossen wird.
Der Schweizer Importeur übernimmt das Umetikettieren oder Umverpacken auf eigene Rechnung, ohne dass ein “Private Label”-Vertrag abgeschlossen wurde
Zwei Optionen sind möglich:
1. Der Schweizer Importeur handelt als “Own Brand Labeller (OBL)”, ein Konzept, das impliziert, dass der OBL das Endprodukt von seinem Lieferanten, in der Regel als “Own Equipment Manufacturer (OEM)” bekannt, geliefert bekommt und daher die Verpackung/Etikettierung so verändert, dass er als derjenige erscheint, der das Produkt auf den Markt bringt.
«OBLs gelten als Hersteller im Sinne der EU-MDR/IVDR.»
Gemäss Frage IV.5 im Guidance Document MDCG 2019-6 (Q&A requirements relating to Notified Bodies) gelten OBLs als Hersteller im Sinne der EU-MDR/IVDR und müssen die entsprechenden Anforderungen für Hersteller erfüllen, was unter anderem bedeutet, dass sie über Folgendes verfügen müssen:
“full and permanent access to the technical documentation; (ability for) post-market surveillance including post market clinical follow-up; sufficient technical competence; and control of the quality system (control of the design, manufacture and/or final verification and testing of the devices).”
2. Der Schweizer Importeur handelt nicht als OBL. Die Tätigkeiten müssen dann im Rahmen von Art. 16 Abs. 2 EU-MDR/IVDR durchgeführt werden. Nach diesem Artikel sind ausschliesslich die folgenden Tätigkeiten erlaubt: die Bereitstellung von Produktinformationen (einschliesslich Zurverfügungstellen von Übersetzungen) und/oder das Umpacken des Produkts, falls dies für die Vermarktung des Produkts in der Schweiz erforderlich ist. Als “erforderlich” betrachtet wird von der MDCG 2021-26 die Notwendigkeit, eine andere Anzahl von Produkten als die vom Hersteller in der Originalverpackung gelieferte Anzahl zu liefern.
In solchen Fällen muss der Schweizer Importeur:
- auf dem Produkt, der Verpackung oder einem Begleitdokument die durchgeführte Tätigkeit angeben,
- über ein Qualitätsmanagementsystem verfügen, das Verfahren zur korrekten Übersetzungen und angemessenen Umetikettierung/Umverpackung umfasst,
- über ein Zertifikat einer benannten Stelle verfügen, das bescheinigt, dass das Qualitätsmanagementsystem den Anforderungen entspricht, und
- Swissmedic und der Hersteller mindestens 28 Tage vor der Bereitstellung des umetikettierten/umverpackten Produkts auf dem Markt über diese Tätigkeiten informieren.
(Art. 16 Abs. 3 und 4 EU-MDR/IVDR sowie Art. 53 Abs. 4 MepV bzw. Art. 46 Abs. 4 IvDV)
Der Schweizer Importeur übernimmt das Umetikettieren oder Umverpacken auf eigene Rechnung mit einem “Private Label”-Vertrag
Obwohl das Konzept der “Private Label” auch in der EU-MDR/IVDR nicht existiert, kann es in der Praxis in gewisser Weise als von Art. 16 Abs. 1 Bst. a abgedeckt angesehen werden. Dies da der Importeur ein Produkt unter seinem eigenen Namen auf dem Markt bereitstellen könnte, wonach der legale Hersteller auf dem Etikett als solcher ausgewiesen wird und für die Erfüllung der Anforderungen an die Hersteller gemäss EU-MDR/IVDR verantwortlich ist.
Diese Möglichkeit ist auch in der Definition des Begriffs “Hersteller” im Rahmen der MepV (und der IvDV) vorgesehen:
“Hersteller: jede natürliche oder juristische Person, die ein Produkt herstellt oder neu aufbereitet oder entwickeln, herstellen oder neu aufbereiten lässt und dieses Produkt unter ihrem eigenen Namen oder ihrer eigenen Marke vermarktet; die in Art. 16 Abs. 1 und 2 der Verordnung (EU) 2017/74511 über Medizinprodukte (EU-MDR) aufgeführten Präzisierungen und Ausnahmen bleiben vorbehalten”
Wie kann Decomplix helfen?
Decomplix berät Sie in allen regulatorischen und qualitätssichernden Fragen im Zusammenhang mit Medizinprodukten, mit besonderem Fokus auf den Schweizer Markt. Wir unterstützen Ihr Unternehmen bei der Suche nach geeigneten Lösungen für Ihre bestehende Vertriebskette und helfen Schweizer Importeuren, sich in den neuen Anforderungen zurechtzufinden und die Einhaltung der Vorschriften auf die effizienteste Weise sicherzustellen.
Darüber hinaus bieten wir seit Mai 2021 Dienstleistungen als Schweizer Bevollmächtigter sowohl unter der MepV als auch unter der IvDV an und vertreten zahlreiche ausländische Hersteller. Diese Dienstleistungen umfassen einen Mandatsvertrag, eine detaillierte Schritt-für-Schritt-Anleitung und Checklisten für Ihr Verständnis der geltenden Anforderungen.
Weiterlesen
- Schweizer Bevollmächtigter für Hersteller von Medizinprodukten
- Welche Anforderungen der MDR gelten für Händler von Medizinprodukten?
- “Legacy”- Medizinprodukte und -IVDs nach EU-Recht
- Sprachliche Anforderungen der EU-MDR — was Hersteller und Distributoren wissen müssen
- Wie die Regulierung von Medizinprodukten die Patientensicherheit erhöht



