
Überwachung von Medizinprodukten nach dem Inverkehrbringen gemäss EU-MDR und IVDR
Die Überwachung nach dem Inverkehrbringen (PMS für Post-Market Surveillance) von CE-gekennzeichneten Medizinprodukten ist kein neues Konzept, und trotzdem verstehen viele Akteure den Zweck und die Anforderungen nicht richtig. Bedenklich ist, dass sich Hersteller vor allem auf die Aktivitäten vor dem Inverkehrbringen konzentrieren und der PMS keine Priorität schenken.
Wenn Sie unsicher sind, welche Schritte es für eine konforme PMS braucht, oder wenn es Sie interessiert, was sich gegenüber den früheren Rechtsvorschriften (MDD, AIMDD, IVDD) geändert hat, lesen Sie weiter.
Ersetzt die Version vom 03.10.2019
Wichtigste Erkenntnisse
- Alle Medizinproduktehersteller müssen ein PMS-System einrichten, das den Anforderungen der EU-MDR oder IVDR entspricht, auch für Legacy-Produkte. Der erste Schritt dazu ist das PMS-Verfahren. Unser Beispiel in diesem Artikel gibt Ihnen eine Anleitung.
- Der PMS-Plan gemäss EU-MDR oder IVDR ist nicht nur eine Tabelle mit Querverweisen auf andere QMS-Verfahren. Er muss auch Methoden, Prozesse und Protokolle erfassen, wie in Anhang III der EU-MDR und der IVDR gefordert.
- Stellen Sie sicher, dass Sie den Leitfaden MDCG 2022-21 gelesen und verstanden haben, bevor Sie mit der Erstellung von PMS-Plänen und PSURs, aber auch PMS-Berichten beginnen.
- PMS-Aktivitäten sind arbeitsintensiv. Stellen Sie sicher, dass Sie über die notwendigen Ressourcen verfügen. Diese können auch ausgelagert werden.
Inhalt
Was ist PMS gemäss EU-MDR und IVDR?
Gemäss den neuen Verordnungen (EU) Nr. 2017/745 über Medizinprodukte (EU-MDR) und 2017/746 über In-vitro-Diagnostika (IVDR) meint “Überwachung nach dem Inverkehrbringen” (PMS für Post-Market Surveillance) den systematischen Prozess, den Hersteller einrichten müssen, um Erfahrungen mit ihren in Verkehr gebrachten Medizinprodukten und IVDs zu sammeln und darauf zu reagieren.
Die Definition von PMS steht in EU-MDR Art. 2 Abs. 60 sowie IVDR Art. 2 Abs. 63:
„Überwachung nach dem Inverkehrbringen“ bezeichnet alle Tätigkeiten, die Hersteller in Zusammenarbeit mit anderen Wirtschaftsakteuren durchführen, um ein Verfahren zur proaktiven Erhebung und Überprüfung von Erfahrungen, die mit den von ihnen in Verkehr gebrachten, auf dem Markt bereitgestellten oder in Betrieb genommenen Produkten gewonnen werden, einzurichten und auf dem neuesten Stand zu halten, mit dem ein etwaiger Bedarf an unverzüglich zu ergreifenden Korrektur- oder Präventivmassnahmen festgestellt werden kann.”
Dies ist keine neue Anforderung. PMS war bereits in den früheren Richtlinien (MDD, AIMDD und IVDD) vorgeschrieben, wenn auch weniger ausdrücklich und ausführlich beschrieben. PMS oder die Sammlung und Überprüfung von Informationen nach dem Inverkehrbringen sind auch in wichtigen internationalen Normen wie ISO 13485 über Qualitätsmanagement-Systeme (QMS) für Medizinprodukte, ISO 14971 über Risikomanagement für Medizinprodukte oder IEC 63204 über das Management des Lebenszyklus von Medizinproduktesoftware vorgeschrieben.
Das PMS-System muss für jedes Produkt unter Berücksichtigung des Produkttyps und seiner Risikoklasse eingerichtet werden und ein integraler Bestandteil des Hersteller-QMS sein. Kurz gesagt, PMS ist ein produktspezifischer Prozess und nicht nur eine Reihe von Dokumenten.
Der PMS-Prozess beinhaltet die Erfassung und Analyse relevanter Daten über Qualität, Leistung und Sicherheit eines Produkts. Diese führen zur Ermittlung, Umsetzung und Überwachung der nötigen Präventiv- und Korrekturmassnahmen. Der PMS-Prozess muss in einem PMS-Plan dokumentiert werden und mündet je nach Risikoklasse in einem PMS-Bericht oder einem “Regelmässig aktualisierter Bericht über die Sicherheit” (PSUR für Periodic Safety Update Report). Siehe auch “Was beinhaltet die PMS gemäss EU-MDR und IVDR?”
Der PMS-Prozess muss während der gesamten Lebensdauer des Produkts kontinuierlich betrieben werden und ist eng mit dem Risikomanagement und den Prozessen der klinischen (Leistungs-) Bewertung verknüpft.
Ist Überwachung nach dem Inverkehrbringen dasselbe wie Marktüberwachung?
Nein.
Die ebenfalls in Kapitel VII von EU-MDR und IVDR beschriebene Marktüberwachung betrifft nicht die Hersteller. Sie bezieht sich auf die von den zuständigen nationalen Behörden durchgeführten Kontrollen der Einhaltung und Durchsetzung der Vorschriften.
Die Marktüberwachung wird nach einem vorher festgelegten Plan durchgeführt und kann Folgendes beinhalten:
- angekündigte und unangekündigte Audits bei Wirtschaftsakteuren,
- Stichproben von Produkten,
- Beschlagnahmung von Produkten, die ein inakzeptables Risiko darstellen oder gefälscht sind.
Die zuständigen Behörden können jederzeit von einem Hersteller, seinem Bevollmächtigten oder Importeur/Händler Produktunterlagen, Informationen oder Muster anfordern.
Die Marktüberwachungspläne werden auf der Grundlage von Risiko- und Vigilanzdaten oder gemeldeten Beschwerden erstellt, was nicht bedeutet, dass Produkte mit geringem Risiko, bei denen keine Zwischenfälle gemeldet wurden, von solchen Inspektionen ausgeschlossen sind. Die zuständige Schweizer Behörde, Swissmedic, führte beispielsweise im März 2023 eine Inspektion bei 27 Herstellern von Klasse-I-Produkten durch.
Welche Medizinprodukte erfordern PMS-Aktivitäten?
Alle in Verkehr gebrachten Medizinprodukte und IVDs unterliegen den PMS-Anforderungen gemäss Kapitel VII der EU-MDR bzw. IVDR.
Dies bedeutet, dass Legacy-Produkte die PMS-Anforderungen gemäss EU-MDR und IVDR anstelle der früheren Richtlinien (MDD, AIMDD und IVDD) erfüllen müssen. Es gelten jedoch folgende Ausnahmen:
- Für Legacy-Medizinprodukte muss gemäss dem Leitliniendokument MDCG 2021-25 das Ergebnis der Überprüfung und Analyse der PMS-Daten nicht zu einer Überarbeitung der technischen Dokumentation gemäss Anhang II und III führen, da Legacy-Produkte keine solche technische Dokumentation benötigen.
- Für Legacy-IVDs wird gemäss dem Leitliniendokument MDCG 2022-8 kein “regelmässig aktualisierter Bericht über die Sicherheit” (PSUR) erwartet, wie es für höhere Risikoklassen in der IVDR verlangt wird. Der PMS-Bericht reicht aus, da die frühere IVDD im Gegensatz zur IVDR keine Risikoklassen vorsah.
In unserem Beratungsalltag stellen wir fest, dass viele “Legacy”-Hersteller ihre PMS-Prozesse noch nicht an EU-MDR oder IVDR angepasst haben und dass die Benannten Stellen die neuen Anforderung offenbar nicht streng genug durchsetzen. Infolgedessen sind die PMS-Pläne und die daraus resultierenden PSUR nicht konform. Eine Umstellung auf die EU-MDR oder IVDR wird dadurch arbeitsintensiver.
Weiter gelten die PMS-Anforderungen der EU-MDR auch für:
- Sonderanfertigungen (CMD), d.h. Produkte, die auf einen bestimmten Patienten zugeschnitten sind. Es gelten jedoch folgende Besonderheiten, da es für jedes CMD nur ein Modell gibt:
- Die PMS-Aktivitäten sind auf Gruppen ähnlicher Produkte anzuwenden (d.h. gleiche Zweckbestimmung, Materialien, verwendete Verfahren, Produktdesign) und nicht auf jedes einzelne CMD.
- Es ist ein Kommunikationskanal zwischen Hersteller und betreffenden Angehörigen der Gesundheitsberufe oder betroffenen Patienten erforderlich, um Rückmeldungen zu Qualität, Leistung und Sicherheit eines bestimmten CMD zu sammeln.
- Für implantierbare CMD der Klasse III (die eine Zertifizierung des Hersteller-QMS durch eine Benannte Stelle erfordern) müssen die PSURs nicht an die Benannte Stelle geschickt werden. Sie müssen lediglich Teil der CMD-Dokumentation sein, die gemäss EU-MDR Anhang XIII erforderlich ist.
- Anhang XVI-Produkte, d.h. Produkte ohne medizinische Zweckbestimmung, die unter die EU-MDR fallen, in Übereinstimmung mit den entsprechenden gemeinsamen Spezifikationen, Verordnung (EU) 2022/2346.
Für Anhang XVI-Produkte der Klassen IIa, IIb und III ist ein PSUR erforderlich, sobald die EU-MDR anwendbar wird. Gemäss den Übergangsbestimmungen in den gemeinsamen Spezifikationen sind dies der:- 31-Dez-2028, wenn keine klinischen Prüfungen geplant sind, oder
- 31-Dez-2029, wenn klinische Prüfungen geplant sind oder laufen.

In unserem Beratungsalltag stellen wir fest, dass viele “Legacy”-Hersteller ihre PMS-Prozesse noch nicht an EU-MDR oder IVDR angepasst haben.
Keinen separaten PMS-Prozess benötigen hingegen Behandlungseinheiten und Systeme, weil diese in Bezug auf die darin enthaltenen Einzelprodukte nicht zusätzlich CE-gekennzeichnet sind, und weil Hersteller von Behandlungseinheiten und Systemen nicht notwendigerweise die legalen Hersteller der Einzelprodukte sind und daher keinen Zugang zu den technischen Unterlagen haben.
Wer ist für die PMS verantwortlich?
Die PMS muss vom Hersteller betrieben werden.
Die PMS-Aktivitäten sind jedoch in Zusammenarbeit mit anderen Wirtschaftsakteuren durchzuführen, da Importeure, Händler und Wiederverkäufer auch in der Lage sind, wichtige Rückmeldungen über das Verhalten des in Verkehr gebrachten Produkts zu geben.
Beim Hersteller muss die “für die Einhaltung der Regulierungsvorschriften verantwortliche Person” (PRRC) die PMS-Aktivitäten überwachen und sicherstellen, dass die Verpflichtungen eingehalten werden.
Für Legacy-Produkte gelten die PMS-Anforderungen zwar auch, ein PRRC ist jedoch nicht erforderlich. Dies wird sowohl aus MDCG 2021-25 für Medizinprodukte als auch MDCG 2022-8 für IVDs ersichtlich.
Was beinhaltet die PMS gemäss EU-MDR und IVDR?
Einfach ausgedrückt, geht es beim PMS-Prozess um das Sammeln, Analysieren, Verarbeiten und Nachverfolgen von PMS-Daten.
Gemäss EU-MDR und IVDR Anhang III Abschnitt 1 Buchstabe a müssen die PMS-Daten Folgendes umfassen:
- Gemeldete Vigilanzfälle, d.h. schwerwiegende Zwischenfälle und Sicherheitskorrekturmassnahmen im Feld,
- nicht schwerwiegende Zwischenfälle und unerwünschte Nebenwirkungen,
- Informationen aus der Vigilanz-Trendberichterstattung,
- sonstige Beschwerden und Rückmeldungen von Anwendern, Händlern/Importeuren, die keine Vigilanzfälle sind,
- wissenschaftliche oder technische Literatur,
- Informationen aus Datenbanken und/oder Registern sowie
- öffentlich verfügbare Informationen über ähnliche Produkte.
Neu müssen gemäss Art. 83 Abs. 4 EU-MDR und Art. 78 Abs. 4 IVDR die zuständigen Behörden und gegebenenfalls die Benannten Stellen über alle Korrektur- oder Präventivmassnahmen (CAPA) informiert werden, die im Laufe der PMS-Aktivitäten getroffen wurden. Diese CAPAs werden in einem zusätzlichen PMS-Datensatz zusammengefasst. Gemäss MDCG 2022-21 entsprechen sie denjenigen CAPAs, die sich beziehen auf:
- Produkte, die bereits auf dem EU-Markt in Verkehr gebracht wurden, d.h. keine CAPA, die während der Entwicklung eröffnet wurden,
- produktbezogene Probleme, die sich auf die Sicherheit, Leistung oder Qualität des Produkts auswirken könnten, einschliesslich QMS-Problemen mit solchen Auswirkungen,
- eine freiwillige Aussetzung des Verkaufs, bei der es sich nicht um eine kommerzielle Entscheidung handelt,
- die Notwendigkeit, neue/geänderte Vorteile und Risiken zu bewerten, die durch PMS-Aktivitäten ermittelt wurden.
Die zu erhebenden PMS-Daten sind sehr umfangreich. Knapp hingegen sind die Fristen, welche die EU-MDR und die IVDR für die Überprüfung der PMS-Daten vorschreiben. Es ist daher wichtig, dass für diesen Überprüfungsprozess genügend Ressourcen bereitgestellt werden, und u.U. PMS-Aktivitäten an Dritte ausgelagert werden. Mehr dazu finden Sie unter “Wie Decomplix helfen kann”.
Wie jeder andere QMS-Prozess muss auch der PMS-Prozess beschreiben, wie die PMS-Aktivitäten vom Hersteller geplant (PMS-Plan), durchgeführt und dokumentiert werden (PMS-Bericht oder PSUR). Siehe auch Kapitel “Wie erstellt man einen PMS-Plan, einen PMS-Bericht oder einen PSUR?”
Laut Art. 83 Abs. 3 EU-MDR bzw. Art. 78 Abs. 3 IVDR sollen die im PMS-Verfahren gesammelten Daten zu folgenden Zwecken verwendet werden:
- Aktualisierung der Nutzen-Risiko-Abwägung und Verbesserung des Risikomanagements gemäss Anhang I Kapitel I;
- Aktualisierung der Auslegung und der Informationen zur Herstellung, der Gebrauchsanweisung und der Kennzeichnung;
- Aktualisierung der klinischen Bewertung;
- Aktualisierung des Kurzberichts über Sicherheit und klinische Leistung gemäss Artikel 32;
- Ermittlung des Bedarfs an Präventiv-, Korrektur- oder Sicherheitskorrekturmaßnahmen im Feld;
- Ermittlung von Möglichkeiten zur Verbesserung der Gebrauchstauglichkeit, der Leistung und der Sicherheit des Produkts;
- gegebenenfalls als Beitrag zur Überwachung anderer Produkte nach dem Inverkehrbringen und
- Erkennung und Meldung von Trends gemäss Artikel 88.
- Die technische Dokumentation wird entsprechend aktualisiert.
Wie die oben genannten Punkte verdeutlichen, muss das PMS-Verfahren daher mit den Verfahren für CAPA, Entwicklung, Herstellung, Risikomanagement, Etikettierung, klinische Bewertung, Leistungsbewertung und technische Dokumentation verknüpft werden.
Folgender Chart zeigt ein Beispiel für die Inputs und Outputs des PMS-Verfahrens.
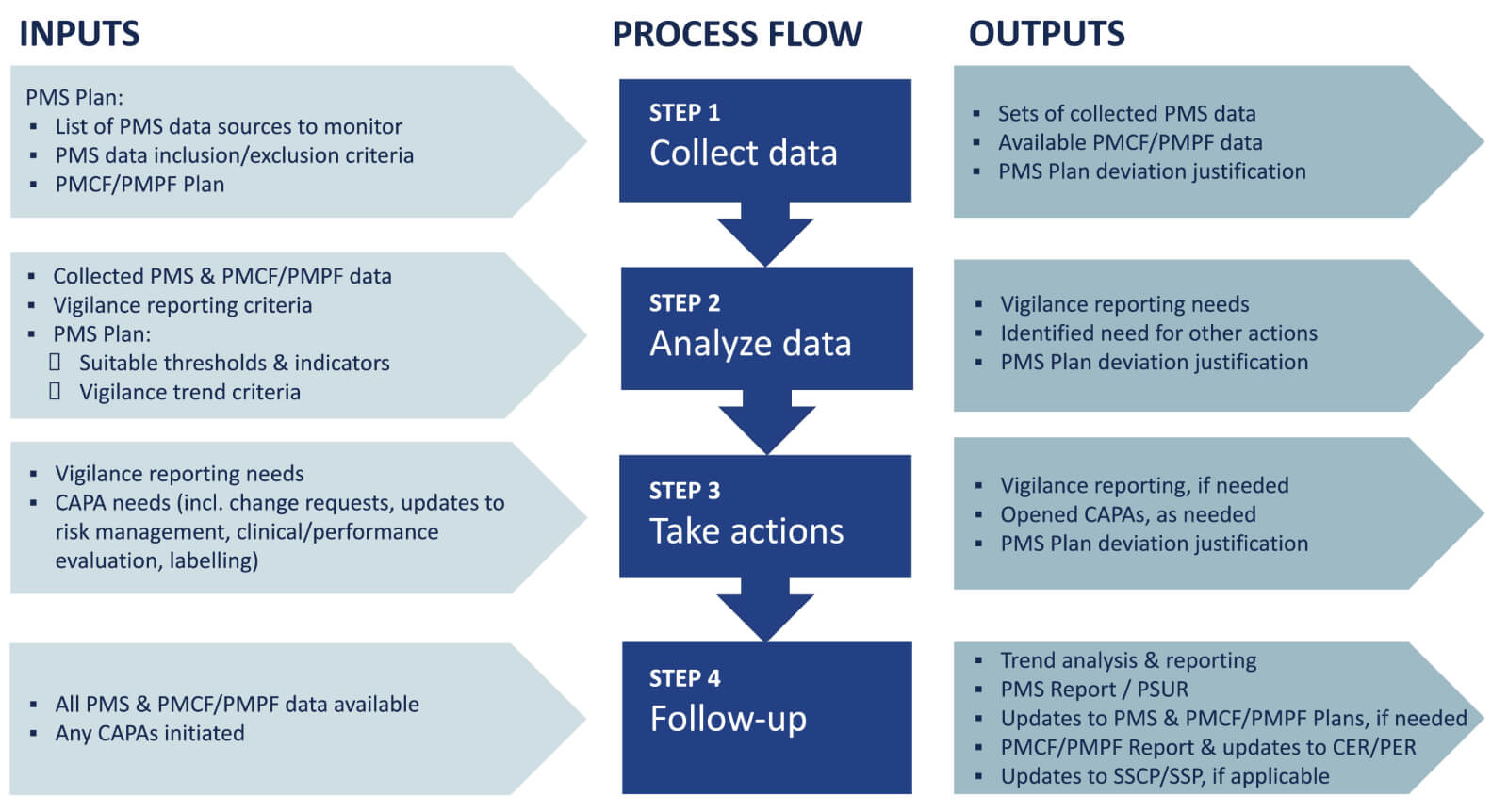
Eine allgemeine Anleitung finden Sie auch im technischen Bericht ISO/TR 20416 über die Überwachung nach dem Inverkehrbringen für Hersteller von Medizinprodukten. Insbesondere die Anhänge enthalten nützliche Beispiele für PMS-Datenquellen, Datenanalysemethoden und PMS-Pläne.
Wie erstellt man einen PMS-Plan, einen PMS-Bericht oder einen PSUR?
Beginnen wir mit dem Ende: dem regelmässig aktualisierten Sicherheitsbericht (PSUR für Periodic Safety Update Report).
Ein PSUR ist immer erforderlich für:
- Medizinprodukte der Klassen Ila, IIb und III, unabhängig davon, ob sie nach EU-MDR oder MDD/AIMDD CE-gekennzeichnet sind.
- IVDs der Klassen C und D, die nach IVDR CE-gekennzeichnet sind.
Die Erstellung von PSURs ist eine wiederkehrende Tätigkeit, die bis zum Ende der Lebensdauer des Produkts erfolgt. Für Medizinprodukte der Klasse IIb/III oder IVDs der Klasse C/D erfolgt sie mindestens jährlich, für Medizinprodukte der Klasse IIa alle zwei Jahre. Die PSURs müssen Teil der technischen Dokumentation für das betreffende Produkt sein, wie in Anhang III der EU-MDR/IVDR festgelegt.
Das Leitliniendokument MDCG 2022-21 schreibt sehr genau vor, wie ein PSUR gemäss EU-MDR zu erstellen ist in Bezug auf Inhalt und Struktur. MDGC 2022-21 kann in Ermangelung einer spezifischen Leitlinie für die IVDR extrapoliert werden.
Anhang I der MDCG 2022-21 enthält die Vorlage für einen PSUR. Die Abbildung unten zeigt ein Beispiel für ein Inhaltsverzeichnis, das sich aus dieser Vorlage ergibt. Die Benannten Stellen erwarten, dass sich Hersteller so eng wie möglich an diese Struktur halten.

Anhang II der MDCG 2022-21 enthält Beispiele für die tabellarische Darstellung von PMS-Daten in einem PSUR. In Anhang III wird erläutert, wie die Datensätze dargestellt werden sollten: pro Basis-UDI-DI, pro Region, von Jahr zu Jahr und unter Verwendung von Begriffen und Codes der IMDRF Adverse Event Terminology (AET).
Die Verwendung der IMDRF-AET-Codes und -Begriffe ist eine Herausforderung, die von vielen Herstellern unterschätzt wird. Die meisten alten Daten sind noch nicht nach diesen Codes kodiert, welche sich zudem laufend verändern können, da die IMDRF-AET-Arbeitsgruppe sie regelmässig überprüft. Hersteller, die sich nicht die Zeit genommen haben, die IMDRF-AET-Codes konsequent in ihre Beschwerdemanagement- und Vigilanzsysteme aufzunehmen, haben es schwer, einen PSUR mit PMS-Daten auszufüllen, die von einer PMS-Überprüfungsperiode zur nächsten vergleichbar sind.

Kurz gesagt, der PMS-Plan ist ein “komprimiertes” Standardverfahren für die PMS, das für ein bestimmtes Produkt oder eine Produktgruppe gilt.
Im Gegensatz zum PSUR gibt es keinen Leitfaden der MDCG für einen PMS-Bericht, der für niedrigere Risikoklassen und für Legacy-IVDs erforderlich ist. Der Inhalt eines PMS-Berichts muss lediglich den Anforderungen von EU-MDR Artikel 85 oder IVDR-Artikel 80 entsprechen, d.h. er beinhaltet eine Zusammenfassung der Ergebnisse und Schlussfolgerungen der Analysen, die sich aus dem PMS-Plan ergeben, sowie eine Begründung und eine Beschreibung der ergriffenen Präventiv- und Korrekturmassnahmen. Die PMS-Berichte können bei Bedarf aktualisiert werden. In der Praxis erstellen die Hersteller PMS-Berichte meist auf der Grundlage von MDCG 2022-21, insbesondere wenn ihr Portfolio auch Produkte höherer Risikoklassen umfasst, die einen PSUR erfordern.
Der PSUR und der PMS-Bericht dienen der Dokumentation der Ergebnisse des PMS-Plans. Gemäss Anhang III der EU-MDR und IVDR muss der PMS-Plan mindestens Folgendes umfassen:
- ein proaktives und systematisches Verfahren für die PMS-Datenerhebung.
- wirksame und geeignete Methoden und Prozesse zur Bewertung der erhobenen Daten
- geeignete Indikatoren und Schwellenwerte für die kontinuierliche Neubewertung der Nutzen-Risiko-Analyse und des Risikomanagements.
- wirksame und geeignete Methoden und Instrumente für die Untersuchung von Beschwerden und die Analyse von marktbezogenen Efahrungen.
- Methoden und Protokolle für die Trendanalyse (d.h. die Feststellung einer statistisch signifikanten Zunahme der Häufigkeit/Schwere von Vorfällen).
- Methoden und Protokolle für eine wirksame Kommunikation mit den zuständigen Behörden, den Benannten Stellen, den Wirtschaftsakteuren und den Anwendern.
- einen Verweis auf die Vigilanz-Meldeverfahren des Herstellers.
- systematische Verfahren zur Ermittlung und Einleitung geeigneter Massnahmen, einschliesslich CAPA.
- Instrumente zur Rückverfolgung und Identifizierung von Produkten, die der CAPA unterliegen.
- ein Plan für klinische Folgemassnahmen nach dem Inverkehrbringen (PMCF) oder für Folgemassnahmen der Leistung nach dem Inverkehrbringen (PMPF) oder eine Begründung, warum ein PMCF/PMPF nicht anwendbar ist.
Kurz gesagt, der PMS-Plan ist ein “komprimiertes” Standardverfahren für die PMS, das für ein bestimmtes Produkt oder eine Produktgruppe gilt.
Als Berater im Bereich CE-Zertifizierung stellen wir fest, dass die meisten Hersteller ihre PMS-Pläne immer noch wie früher erstellen, was für EU-MDR und IVDR nicht mehr ausreicht.
Obwohl die obige Liste explizit ist und nur einmal einen Verweis erwähnt, erstellen Hersteller ihre PMS-Pläne immer noch mit einer einfachen Tabelle mit Verweisen auf QMS-Verfahren und Arbeitsanweisungen, ohne Details zu Indikatoren und Schwellenwerten zur Ergreifung von Massnahmen. Zudem liefern sie oft ungültige Begründungen, warum der PMCF/PMPF nicht anwendbar sei.
Die einfachste und konformste Art, einen PMS-Plan zu erstellen, ist das “Reverse Engineering” von MDCG 2022-21: Jedes Element, das in einen PSUR aufgenommen werden muss, ist Teil des entsprechenden PMS-Plans.
Was sind “geeignete Schwellenwerte und Indikatoren”?
Es ist ein weit verbreiteter Irrtum, dass die PMS-Schwellenwerte und -Indikatoren die gleichen sind wie die Auslöser für die Vigilanz-Trendmeldung. Obwohl es gewisse Überschneidungen gibt, sind sie nicht dasselbe und werden in Anhang III Abschnitt 1(b) von EU-MDR und IVDR in verschiedenen Unterabsätzen erwähnt.
Der Umfang der Vigilanz-Trenderfassung ist auf nicht-schwerwiegende Vorfälle beschränkt, um Trends zu erkennen, die gemäss EU-MDR Art. 88 oder IVDR Art. 83 gemeldet werden müssen. Gemeint sind Trends, die erhebliche Auswirkungen auf das Nutzen-Risiko-Profil des Produkts haben und zu inakzeptablen Risiken geführt haben oder führen können. Dies gilt bei Medizinprodukten auch für erwartete unerwünschte Nebenwirkungen und bei IVDs für erwartete fehlerhafte Ergebnisse.
Der Hersteller muss deren signifikante Zunahme feststellen, indem er die Daten innerhalb eines bestimmten Zeitraums (in der Regel der PMS-Überprüfungszeitraum) mit der vorhersehbaren Häufigkeit oder Schwere solcher Vorfälle vergleicht. Die Methodik zur Bestimmung von Trends muss im PMS-Plan beschrieben werden. Eine Methodik dürfte Teil eines künftigen Leitfadens der MDCG sein. In der Zwischenzeit erweist sich Anhang C des alten Leitfadens der Global Harmonization Task Force, GHTF/SF2/N36R7, als sehr hilfreich.
Der Umfang der im PMS-Plan zu beschreibenden PMS-Indikatoren und -Schwellenwerte ist breiter gefasst, da diese für alle Datensätze benötigt werden, um das Nutzen-Risiko-Profil kontinuierlich neu zu bewerten und etwaigen Handlungsbedarf zu ermitteln. Die Indikatoren und Schwellenwerte müssen für die Art des analysierten PMS-Datensatzes aussagekräftig sein und müssen den aktuellen PMS-Überprüfungszeitraum mit den vorangegangenen Zeiträumen für einen Gesamtzeitraum von vier Jahren in Übereinstimmung mit der MDCG 2022-21 vergleichen. Die Methoden können quantitativ (z.B. die statistische Analyse, die zur Identifizierung von Vigilanztrends erforderlich ist), qualitativ (z.B. die Erkennung von Verschiebungen im Stand der Technik oder neuer Risiken in der durchgesehenen wissenschaftlichen Literatur) oder halbquantitativ (z.B. die Meta-Analyse veröffentlichter Studien, bei der qualitative Sortierung und quantitative Analyse kombiniert werden) sein. Der technische Bericht ISO/TR 20416 enthält einige Hinweise für die Auswahl geeigneter Methoden.
Was sind die Unterschiede zwischen PMS und PMCF/PMPF?
PMCF steht für “Post-market clinical Follow-up” – auf Deutsch “klinische Nachbeobachtung nach dem Inverkehrbringen”. PMPF steht für “Post-market performance Follow-up” – auf Deutsch “Leistungsstudien nach dem Inverkehrbringen”.
PMCF gemäss EU-MDR und PMPF gemäss IVDR beziehen sich auf den Prozess, der für die kontinuierliche Aktualisierung der klinischen Bewertung (für Medizinprodukte) bzw. der Leistungsbewertung (für IVDs) erforderlich ist.
Sowohl die EU-MDR (in Anhang XIV, Teil B, Punkt 5) als auch die IVDR (in Anhang XIII, Teil B, Punkt 4) verlangen, dass der PMCF/PMPF “im Plan des Herstellers zur Überwachung nach dem Inverkehrbringen behandelt wird”. Kurz gesagt, der PMCF- oder PMPF-Plan muss Teil des entsprechenden PMS-Plans sein, und es wird erwartet, dass eine Zusammenfassung des PMCF/PMPF-Berichts in den PSUR integriert wird.
Einige der PMCF/PMPF-Daten, die so genannten “general” PMCF/PMPF, überschneiden sich mit den PMS-Daten, wie in der folgenden Abbildung dargestellt.
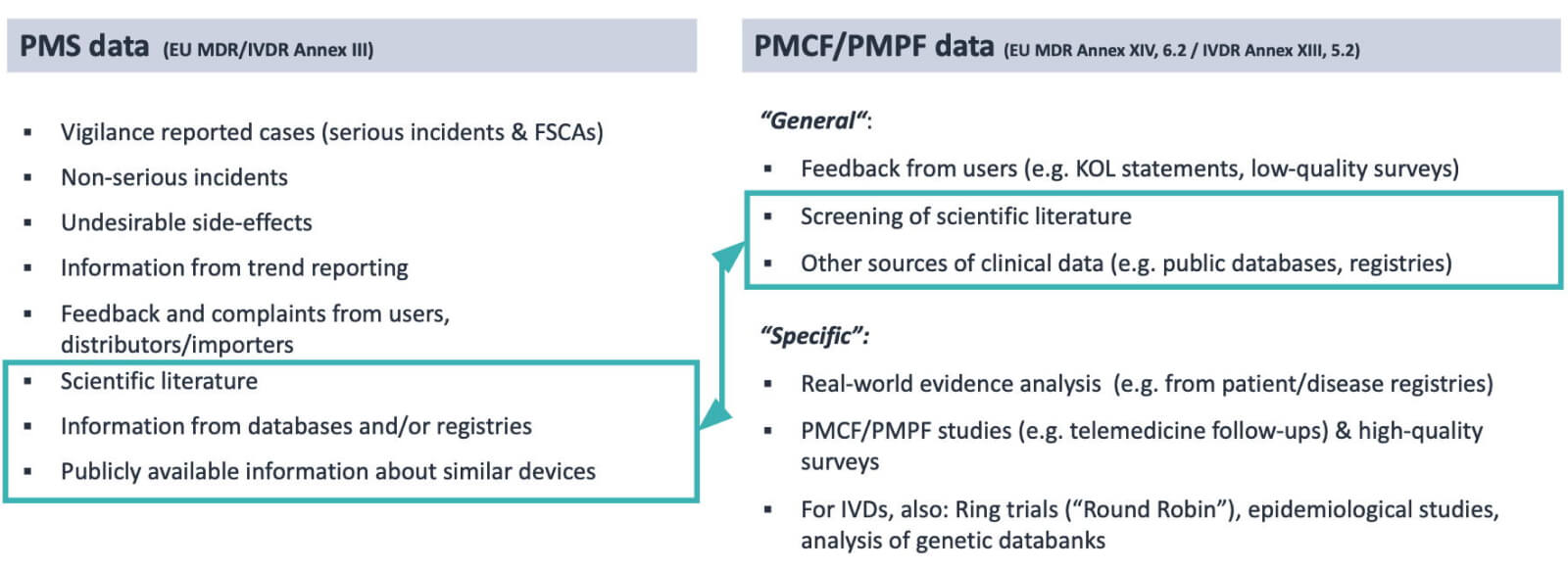
Da der PMCF/PMPF die proaktive Sammlung aller klinischen Daten und Leistungsdaten ist, um die Klinische Bewertung (CER) oder Leistungsbewertung (PER) auf dem neuesten Stand zu halten, ist es sinnvoll, die gemeinsamen, sich überschneidenden Daten als PMCF/PMPF-Daten und nicht als PMS-Daten zu melden.
Wie Decomplix helfen kann
Decomplix bietet Beratungsdienste in allen regulatorischen und qualitätssichernden Fragen im Zusammenhang mit Medizinprodukten und IVDs. Wir können Ihr Unternehmen dabei unterstützen, alle PMS- und PMCF/PMPF-Anforderungen zu erfüllen und die Einhaltung der Vorschriften auf die effizienteste Weise sicherzustellen. Wenn Sie Ihre Bedürfnisse mit uns besprechen möchten, kontaktieren Sie uns bitte für ein unverbindliches Angebot zur CE-Kennzeichnung.
Weiterlesen
- Qualitätsmanagement-System nach ISO 13485 zertifizieren
- Risikomanagement für Medizinprodukte unter EU-MDR und ISO 14971
- Medizinproduktesoftware (MDSW) gemäss EU-MDR und IVDR
- “Legacy”-Medizinprodukte und -IVDs nach EU-Recht
- Anhang XVI und EU-MDR — Leitfaden zur Regulierung von Produkten ohne medizinische Zweckbestimmung



