
UDI for Medical Device Software (MDSW) under EU MDR
In this blog, we summarize what medical device software (MDSW) manufacturers need to do in terms of UDI under Regulation (EU) No. 2017/745 on medical devices from a practical standpoint, in as much detail and as simply as possible.
You have a mobile app or a webtool that qualifies as a medical device under Regulation (EU) No. 2017/745 on medical devices (EU MDR) and do not understand how to implement the Unique Device Identifier (UDI) requirements for software only? You have read the EU MDCG guidance documents on UDI, the EU Commission factsheet on UDI, browsed the new EU UDI Helpdesk, and are still unsure about how to proceed? Here is everything you need to know.
Content:
What is UDI?
Unique Device Identifier (UDI) is a system intended to unambiguously identify medical devices and to allow rapid access to device related information.
Although medical device UDI requirements are already in place or being developed in variable forms in many jurisdictions (e.g. USA, China, Brazil, South Korea, Saudi Arabia), there is no harmonized or universal application of UDIs and it is essential to verify the requirements in each geography of interest for the manufacturer. The present blog refers only to UDI requirements that apply to medical devices CE-marked under the EU MDR, as they apply to the manufacturer. The requirements do not apply to custom-made and investigational devices. And, specifically for software, only software that is commercially available on its own and software that constitutes a medical device in itself (i.e. medical device software, MDSW) shall be subject to these requirements.
There are 3 aspects of the UDI to be considered by the manufacturer, in chronological order:
- The UDI code, to be assigned (and maintained) by the manufacturer for any medical device CE-marked under the EU MDR. The obligation to assign UDI codes started from the EU MDR’s date of application, 26-May-2021.
- The data associated with a UDI code, to be uploaded (and maintained) by the manufacturer in the EUDAMED database. This entails the initial registration and any updates to information and/or registration of additional UDIs.
- The UDI carrier, i.e. Automated Identification for Data Capture (AIDC) and/or human-readable interpretation (HRI) representation of the UDI, to be placed by the manufacturer on the device labelling and, in some cases, on the device itself. Different timelines for UDI carriers are mandated per device risk class.
What UDI codes exist under EU MDR?
The UDI code is a unique, alphanumeric code, which consists of two parts:
- UDI Device Identifier (UDI-DI): A static code specific to a version or model of a medical device. The UDI-DI is the identifier used to access the UDI module of the EUDAMED database.
- UDI Production Identifier (UDI-PI): Different variable (dynamic) codes related to aspects of the production unit. A medical device can contain various UDI-PIs (e.g. serial/lot number, manufacturing date or expiry date). For MDSW, the means of manufacture control corresponds to software identification. The UDI-PIs together with the UDI-DI shall appear on the device labels and the device itself, which, in the case of MDSW, could be elements of the user interface, and are not part of the data that needs to be registered in the EUDAMED database.
In addition, a Basic UDI-DI (BUDI) is required under the EU MDR. What is it?
Also a static code, like the UDI-DI, the BUDI connects MDSW from the same manufacturer (same Single Registration Number, SRN) that have the same intended purpose, the same risk class, and same essential design and manufacturing characteristics. The BUDI can be viewed as the “family” code. The purpose of the BUDI is to be the main access key for device-related information in the EUDAMED database and in relevant device documentation, i.e. EU product certificates issued by Notified Bodies, EU Declaration of Conformity, Technical Documentation, Summary of Safety and Clinical Performance, Vigilance and Post-Market Surveillance reports, Free Sales certificates. Per guidance document MDCG 2018-1, recently updated, a UDI-DI shall be associated with only one BUDI. Moreover, a BUDI cannot be referenced in different EU product certificates and should not be referenced in different sets of Technical Documentation or different EU Declarations of Conformity. The BUDI is not required to appear on the device itself or its labeling.
What are the “core data elements” associated with the UDI?
Under the EU MDR, the manufacturer shall provide and continuously maintain up-to-date the following data in the EUDAMED database:
- Information about its role and general information on the devices placed on the market, per EU MDR Annex VI, Part A, and
- The so-called “core data elements”, a number of given data fields in EUDAMED, to be provided together with the UDI-DI, per EU MDR Annex VI, Part B.
The format and definition of these UDI “core data elements” are provided in document UDIWG 2018-1. An excerpt of those most relevant data fields for MDSW is presented below.
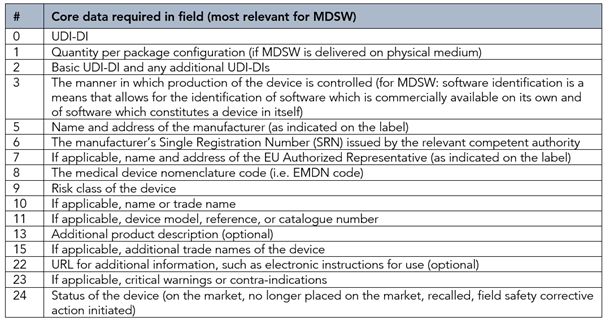
Most relevant core data fields for MDSW
Now, if your MDSW is part of a System or Procedure Pack, as defined in EU MDR Article 2 (10) and (11), respectively, information should be added for the core data elements at the system level, namely:
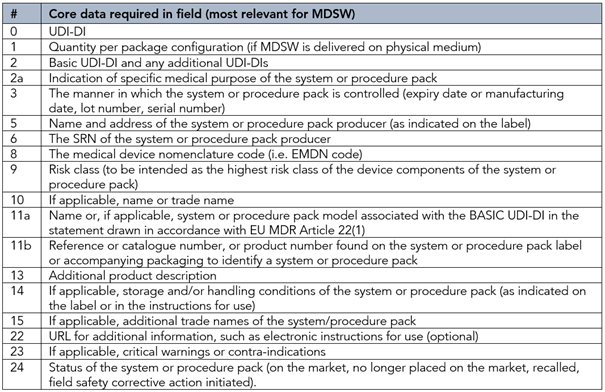
Additional information for MDSW
In practice, what do you need to do?
If you have not started, make UDI a priority. The obligation for UDI assignment for non-legacy devices applies from the EU MDR’s date of application, i.e. 26 May 2021.
The following steps will help you deploy your UDI project for MDSW.
1. Register with a EU-accredited agency for UDI assignment
UDI codes must be issued under the rules of a EU-accredited assigning agency. Following a call for applications launched at the end of 2018, the Commission designated the following entities, per Implementing Decision (EU) 2019/939:
- GS1 AISBL
- Health Industry Business Communications Council (HIBCC)
- International Council for Commonality in Blood Banking Automation (ICCBBA)
- Informationsstelle für Arzneispezialitäten GmbH (IFA)
The table below provides an overview of the type of UDI activities and geographic scope for each of these issuing entities.
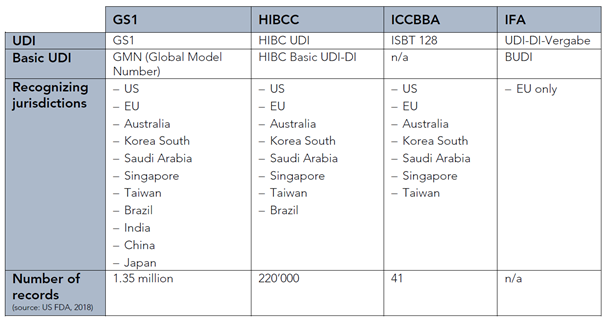
Types of UDI activities
Once registered with your agency of choice, you will be granted the company prefix, e.g. GS1 Global Company Prefix (GCP), the individual identification key that gives you access to the system. In case of changes in the legal status of your company (e.g. name change, merger, acquisition, spin-off), you must notify the agency.
2. Set up a UDI Management procedure
The manufacturer’s Quality Management System (QMS) should include a UDI Management procedure encompassing at a minimum the following aspects:
- The structure of the manufacturer’s UDI-DIs and UDI-PIs.
- The process and criteria for grouping MDSW under a BUDI.
- UDI-PI labelling process (unless described in the general device labelling procedure). In addition, if your MDSW is delivered on a physical medium (e.g. CD, USB-key), the IT and logistics aspects of barcodes will need to be thoroughly considered.
- UDI-DI and BUDI change management process, i.e. criteria triggering a new code per EU MDR Annex VI Part C and MDCG 2019-5 (unless described in the general change management procedure).
- UDI data master maintenance (process for establishing and keeping up to date the list of all BUDIs, UDI-DIs and their associated core data). The actual list of UDI-DIs shall be part of the MDSW’s Technical Documentation, as required by EU MDR Article 27(7).
- The process for electronic transmission and consolidation of UDI data in EUDAMED and/or in any bespoke national databases (for example, in Switzerland).
Unless your company has experience with GTIN management, it is recommended that you appoint a dedicated individual, with regulatory and technical knowledge, to oversee the UDI process throughout the device lifecycle, particularly if your MDSW is provided on a physical medium (e.g. CD, USB key) and AIDC needs to be implemented.
3. Assign the individual UDI-DIs
The UDI-DI shall be assigned by the manufacturer at the software system level, for MDSW that is commercially available on its own. If provided in a physical medium (e.g. CD or USB key), each level of packaging (excluding shipping containers) shall be assigned its own UDI-DI. GS1’s Global Trade Item Number (GTIN) corresponds to the UDI-DI. The structure of the UDI-DI includes the company prefix, e.g. GS1 Global Company Prefix (GCP), the manufacturer’s internal code for the product (e.g. model number), and ends with 1 check digit that is automatically calculated by the system.
An example of UDI-DI structure, built through the GS1 system is shown below.
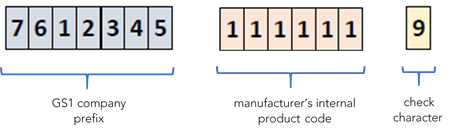
Example of UDI-DI structure based on GS1 system
EU MDR Annex VI, Part C, sections 3 and 6 shall be considered when assigning/updating all identification numbers. In addition, any relevant standards and guidelines of the UDI issuing agency chosen would need to be followed.
4. Assign the Basic UDI-DI (BUDI)
If you only have 1 MDSW variant or a few variants, grouping their UDI-DIs on the basis of BUDI criteria (same risk class, same intended purpose, and same design/manufacturing characteristics) is a no-brainer. Just bear in mind that a UDI-DI (GTIN) shall not be used as a replacement for the Basic UDI-DI (GMN). Grouping diverse MDSW (different risk classes or intended purposes or software architecture/configuration) under different BUDIs needs to be carefully considered and documented, bearing in mind plans for future MDSW extensions or feature additions. If your MDSW products were already CE-marked under the previous Directive 93/42/EEC on medical devices (MDD), you have certainly already realized that the medical device families/grouping used for CE-marking under the MDD might not be suitable device groups for BUDI criteria. MedTech Europe’s Guidance on Basic UDI-DI Assignment provides useful tips on how to make grouping decisions. Once you have decided the necessary BUDIs for your various MDSW, you can build the corresponding codes. A BUDI code assigned by the manufacturer shall have maximum 25 characters (the maximum length of the UDI-DI as established by the issuing entities). Its structure includes the company prefix, e.g. GS1 Global Company Prefix (GCP) and the manufacturer’s internal code for the device group (which can be numeric or alphanumeric, and is case-sensitive), and it incorporate a 2-digit check character, based on an algorithm defined by the issuing entity.
An example of BUDI structure, built through the GS1 system is shown below:
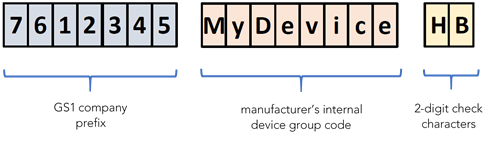
Example of Basic UDI-DI (BUDI) structure based on GS1 system
5. Build the UDI-PIs
The UDI-PIs are numeric or alphanumeric codes used to label the unit of device production. Basically, they correspond to the serial number or lot number (depending on the “manufacturing control mechanism” applied for a given type of medical device by the manufacturer), with additional information on manufacturing or expiry dates. They allow the unambiguous identification of each device unit. The Application Identifiers (AI) of the issuing agency are used to generate the UDI-PIs. For MDSW, the “manufacturing control mechanism” is considered to be the software identification and shall be part of the UDI-PI, per EU MDR, Annex VI, Part C, section 6.5.1. This means that the AI for batch or lot number would be used in the MDSW UDI-PI preceding the software release version. If provided in a physical medium (e.g. CD or USB key), UDI-PIs are also required on the labels. The example below shows the UDI-DI and UDI-PIs represented in 1D (linear) barcode and a 2D (data matrix) barcode, both readable with a barcode scanner, as well as in human-readable format (concatenating the UDI-DI and UDI-PIs parts, preceded by the corresponding GS1’s AIs).

Example of UDI-DI and UDI-PIs represented in GS1 linear and DataMatrix barcodes
For details on the AIs from each UDI issuing entity, refer to Appendix A of IMDRF’s UDI WG applica-tion guide N48 FINAL: 2019.
6. Register and upload UDI data in EUDAMED
As a preliminary step to product registration in EUDAMED, the manufacturer (and also any producer of Procedure Packs or Systems) needs to complete the Economic Operator registration in the corresponding module of the EUDAMED database and obtain the Single Registration Number (SRN) for its role under the EU MDR. The process for this registration, possible since December 2020, is detailed in the corresponding EU’s Actor Registration Module webpage. Per EU MDR Art. 123 (d), actor registration must be completed 6 months after full availability of EUDAMED, which is now planned for May 2022. The SRN uniquely identifies a company’s role and it is assigned by the national competent authority upon the company’s registration request in EUDAMED. The SRN structure includes the ISO code for the country where the company is established, the Economic Operator’s role abbreviation (i.e. “MF” for manufacturer, “PR” for Procedure Pack or System producer, “AR” for EU Authorized Representative, or “IM” for EU importer), and a 9-digit company identification.
A SRN example for a German manufacturer is shown below:
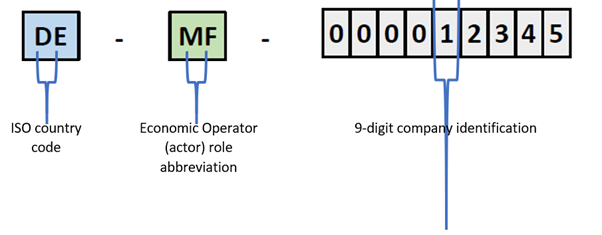
Example of Single Registration Number (SRN) in EUDAMED
For companies established outside the EEA, the EU Authorized Representative is responsible for verifying the company’s registration request prior to submitting it to the national competent authority. This would apply to Swiss manufacturers and producers of Procedure Packs or Systems, since Switzerland is considered a third country for the effects of the EU MDR since 26 May 2021. A parallel process for assigning Swiss Registration Numbers (CHRN) to Economic Operators in Switzerland has been established by the Swiss competent authority, Swissmedic, per Article 55 of the revised Swiss Medical Device Ordinance (MedDO, SR 812.213).
Product registration in the UDI module of the EUDAMED database is expected to be possible from September 2021. After that, manufacturers will have until May 2024 to register the products (UDI-DI and BUDI) in EUDAMED, per EU MDR Article 123(e), i.e. 18 months after the date of Article 123(d). At the time of compiling this blog post, it is still unclear how this will apply in Switzerland. A parallel registration system for devices shall be implemented by the Swiss competent authority, Swissmedic, to gather information on UDIs of the devices placed on the Swiss market. For product registration in EUDAMED, you will need the BUDI and all UDI-DIs generated as well as the associated core data elements, per EU MDR Annex VI, Part A. Guidance document MDCG 2019-4 on the timelines for EUDAMED registration of data elements provides more information. It is important to highlight that, the full registration of devices will be a prerequisite for the notification of serious incidents or Field Safety Corrective Actions (FSCA) in EUDAMED.
After the initial product registration, you will have to maintain continuously up-to-date the UDI information in EUDAMED and to register new devices (i.e. new UDI-DI with or without new BUDI) prior to launch. Any changes made to a core data element, which does not require a new UDI-DI, shall be updated within 30 days of the change being made.
7. Select an appropriate UDI carrier, if needed
The obligation to place the UDI carrier on device labeling under EU MDR applies in phases, according to the following timelines:
|
Action |
Class III & implantable devices |
Class IIb & Class IIa devices |
Class I devices |
|
Placing UDI carriers on device labels, per EU MDR Art. 123(3)(f) |
26-May-2021 |
26-May-2023 |
26-May-2025 |
|
Direct marking of reusable devices, per EU MDR Art. 123(3)(g) |
26-May-2023 |
26-May-2025 |
26-May-2027 |
UDI placement criteria for software are clearly established in EU MDR Annex VI, Part C, point 6.5.4. Three situations are possible:
- For MDSW delivered on a physical medium (e.g. CD, USB key), each packaging level shall bear both the human-readable and machine-readable (i.e. AIDC) representation of the complete UDI-PI. The UDI-PI applied to the physical medium containing the MDSW and its packaging shall be identical to the UDI-PI assigned to the system level software.
- For MDSW not delivered on a physical medium but having a user interface (e.g. mobile apps, webtools), only the human-readable interpretation (HRI) of the UDI-PI shall be required. It shall be provided on a readily accessible screen for the user in an easily-readable plain-text format (e.g. on the ‘about’ file, splash screens, or included on the start-up screen).
- For MDSW lacking a user interface (e.g. middleware for image conversion), the human-readable representation of the UDI-PI shall be conveyed through an application programming interface (API).
In either case, the human-readable format of the UDI-PI shall include the Application Identifiers (AI) for the standard used by the issuing entity, so as to assist the user in identifying the UDI and determining which standard is being used to create the UDI.
For MDSW delivered on a physical medium, the UDI-PI code should be on each applicable packaging level, from the unit of use to the highest package level, except logistics units. It is important to highlight that the implementation of a UDI-PI carrier constitutes a complex project for a company and that it can only be successfully achieved when implemented in the company’s Enterprise Resource Planning (ERP) system and with dedicated IT and logistic resources. The costs and timelines of such a project need to be assessed as early as possible and might lead to a decision to change the MDSW delivery method, from physical to online medium. Although not a EU MDR requirement, in case MDSW is delivered on a physical medium, thus requiring a barcode, verification of the barcode’s print quality is recommended to help ensure readability throughout the supply chain. To that end, standard series ISO 15415 provide relevant requirements for 2D and linear barcodes.
8. Maintain UDIs throughout the MDSW lifecycle
As part of the manufacturer’s change management process, software changes shall be assessed in consideration of the criteria for generating a new UDI-DI set forth in EU MDR Annex VI, Part C, sections 6.5.2 and 6.5.3 as well as in guidance document MDCG 2018-5 on UDI assignment to medical software. EU MDR Annex VI, Part C, Section 6.5.2 requires a new UDI-DI for software “whenever there is a modification that changes:
- the original performance,
- the safety or the intended use of the software
- the interpretation of data.
Such modifications include new or modified algorithms, database structures, operating platforms, architecture, user interfaces and new channels for interoperability.”
And in accordance with EU MDR Annex VI, Part C, point 6.5.3, minor software revisions (which would only require a new UDI-PI and not a new UDI-DI) are considered to be those “generally associated with bug fixes, usability enhancements that are not for safety purposes, security patches or operating efficiency. Minor software revisions shall be identified by a defined manufacturer-specific form of identification.”
Per guidance document MDCG 2018-1, a new UDI-DI shall be required whenever there is a change that could lead to misidentification of the device and/or ambiguity in its traceability. This is further elaborated for MDSW in guidance document MDCG 2018-5, which considers that, in order to guarantee the traceability and correct identification of MDSW, the following types of software changes would require a new UDI-DI:
- Any change of the Basic UDI-DI.
- Any changes which impact the original performance, safety, or the interpretation of data.
- A change to the name or trade name, version or model number, critical warnings or contra-indications, user interface language.
What about “legacy devices”?
“Legacy devices”, i.e. medical devices that remain in conformity with Directive 93/42/EEC (MDD) after the EU MDR’s date of application, are not subject to UDI obligations but they should be registered in EUDAMED. To that end, the EU Commission has decided to use a Eudamed-DI to be assigned to the device instead of the Basic UDI-DI and a Eudamed-ID to be assigned instead of the UDI-DI. The Eudamed DI could be either entirely generated by EUDAMED or the manufacturer could partly assign the DI code. And the Eudamed ID will be automatically and fully generated by EUDAMED, from the Eudamed DI code. Timelines for EUDAMED registration as described above also apply to legacy devices.
More information on the operational aspects of the registration of “legacy devices” is available in the guidance document MDCG 2019-5 on UDI for legacy devices. Alternatively, we recommend our blog article on the topic of “Legacy Devices”.
How can Decomplix help medical software manufacturers?
Decomplix provides assistance in each step of the medical CE certification process for product manufacturers, including medical software. You can learn more about our services here.



