
UDI für Medizinprodukte-Software unter der EU-MDR
In diesem Blog zeigen wir aus einer praktischen Perspektive, was Hersteller von medizinischer Software (medical device Software MDSW) in Bezug auf die Verordnung (EU) 2017/745 über Medizinprodukte (EU-MDR) beachten müssen – so einfach wie möglich und so detailliert wie nötig.
Sie haben das EU MDCG Guidance-Dokument zu UDI gelesen, das Factsheet der EU Kommission studiert und den EU UDI Helpdesk besucht und wissen immer noch nicht, was Sie mit Ihrer App oder Ihrem Webtool umsetzen müssen? Für mobile Apps oder Webtools, die unter der Verordnung (EU) 2017/745 über Medizinprodukte (EU-MDR) als Medizinprodukt gelten, müssen UDI-Anforderungen implementiert werden.
Inhalt:
Was ist UDI?
Unique Device Identifier (UDI) ist ein System, welches Medizinprodukte eindeutig identifizieren und einfachen Zugang zu Informationen zum Produkt ermöglichen soll. International haben viele Länder die Implementation von UDI bereits in ihre Medizinproduktregulierung aufgenommen (beispielsweise USA, China, Brasilien, Südkorea, Saudi Arabien), es findet sich jedoch kein harmonisierter oder universeller Einsatz von UDI. Für Hersteller ist es deshalb zentral, die Anforderungen an UDI länderspezifisch zu betrachten. Dieser Blog setzt sich ausschliesslich mit den UDI-Anforderungen auseinander, welche für Hersteller von CE-zertifizierten Medizinprodukten unter MDR gelten. Für Spezialanfertigungen und Prüfprodukte sind diese nicht anwendbar. Nur Software, welche im Handel eigenständig erhältlich ist sowie Software, die ein Medizinprodukt an sich darstellt (d.h. Medizinische Software MDSW) unterliegen diesen Anforderungen.
Die folgenden drei Aspekte von UDI müssen von Herstellern in chronologischer Reihenfolge beachtet werden:
- Der UDI-Code, welcher vom Hersteller für jedes nach EU-MDR CE-gekennzeichnete Medizinprodukt vergeben (und verwaltet) werden muss. Die Pflicht zur Vergabe von UDI-Codes setzt mit dem Anwendungsdatum der EU-MDR am 26. Mai 2021 ein.
- Die mit einem UDI-Code verknüpften Daten, die vom Hersteller in die Datenbank EUDAMED hochgeladen (und gepflegt) werden müssen. Dies umfasst die Erstregistrierung und sämtliche Aktualisierungen der Informationen und/oder die Registrierung zusätzlicher UDIs.
- Der UDI-Träger, d.h. die automatische Identifikation und Datenerfassung (Automatic Identification and Data Capture AIDC) und/oder die vom Menschen lesbare Form (Human Readable Interpretation HRI) des UDI, ist vom Hersteller auf der Produktkennzeichnung und in gewissen Fällen auch auf dem Produkt selbst anzubringen. Für die UDI-Träger gelten je nach Risikoklasse des Produkts unterschiedliche Fristen.
Welche UDI-Codes gibt es unter der EU-MDR?
Der UDI-Code ist ein eindeutiger, alphanumerischer Code, welcher aus zwei Teilen besteht:
- UDI-Produktkennung (UDI Device Identifier UDI-DI): ein statischer Code spezifisch für eine Version oder ein Modell eines Medizinprodukts. Der UDI-DI ist die Kennung, welcher für den Zugriff auf das UDI-Modul von EUDAMED verwendet wird.
- UDI-Herstellungskennung (UDI Production Identifier UDI-PI): Verschiedene variable (dynamische) Codes, die sich auf Aspekte der Produktionseinheit beziehen. Ein Medizinprodukt kann verschiedene UDI-PIs enthalten (z.B. Serien-/Losnummer, Herstellungs- oder Ablaufdatum). Bei MDSW entspricht die Herstellungskontrolle der Software-Identifikation. Die UDI-PIs zusammen mit dem UDI-DI sollen auf dem Produktelabel und dem Produkt selbst angebracht sein. Im Falle von MDSW können dies Elemente der Benutzeroberfläche sein und gehören nicht zu den Daten, welche in EUDAMED registriert werden müssen.
Zusätzlich ist unter der EU-MDR eine Basis-UDI-DI (Basic UDI-DI BUDI) erforderlich. Was ist das?
Die BUDI ist wie die UDI-DI ein statischer Code, welcher die MDSW vom gleichen Hersteller (gleiche einmalige Registrierungsnummer (Single Registration Number SRN)), die den gleichen Verwendungszweck, die gleiche Risikoklasse sowie die gleichen wesentlichen Design- und Herstellungsmerkmale aufweisen. Die BUDI kann als „Familien-Code“ betrachtet werden. Die BUDI bildet den Hauptzugriff für gerätebezogene Informationen in EUDAMED und in der Produktdokumentation, d.h. von Benannten Stellen ausgestellte EU-Produktzertifikate, EU-Konformitätserklärungen, technische Dokumentation, Kurzbericht über Sicherheit und klinische Leistung, Reports zur Vigilanz und Überwachung nach dem Inverkehrbringen und Freiverkaufszertifikate (FSCs). Gemäss der Guidance MDCG-2018-1, welche kürzlich aktualisiert wurde, soll ein UDI-DI nur mit einer BUDI verknüpft werden. Zudem kann eine BUDI nicht in verschiedenen EU-Produktzertifikaten referenziert werden und sollte auch nicht in verschiedenen Datensätzen der technischen Dokumentation oder verschiedenen EU-Konformitätserklärungen verwendet werden. Die BUDI muss nicht auf dem Produkt oder seinem Label angegeben werden.
Was sind die mit dem UDI verbundenen „zentralen Datenelemente“?
Gemäss EU-MDR muss der Hersteller die folgenden Daten in EUDAMED bereitstellen und kontinuierlich aktualisieren:
- Informationen über seine Rolle und allgemeine Informationen über die in Verkehr gebrachten Produkte, gemäss EU-MDR Anhang VI, Teil A.
- Die sogenannten „zentralen Datenelemente“, eine Anzahl vorgegebener Datenfelder in EUDAMED, welche zusammen mit der UDI-DI anzugeben sind gemäss EU-MDR Anhang VI, Teil B.
Das Format und die Definition dieser „zentralen Datenelemente“ werden im Dokument UDIWG-2018-1 erläutert. Im Folgenden werden die für MDSW wichtigsten Datenfelder aufgelistet:
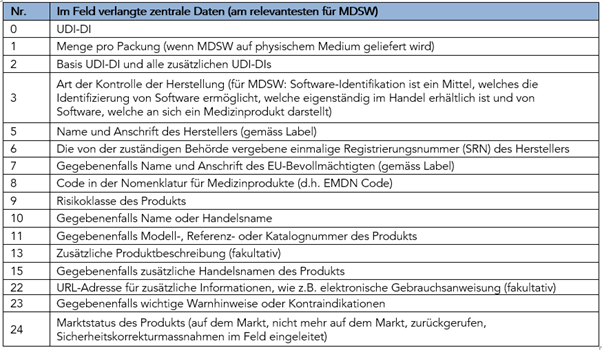
Die wichtigsten Datenfelder für MDSW
Wenn es sich bei der MDSW um einen Teil eines Systems oder einer Behandlungseinheit gemäss EU-MDR Artikel 2 (10) und (11) handelt, sollten Informationen für die zentralen Datenelemente auf Systemebene hinzugefügt werden:

Zusätzliche Informationen für MDSW
Was bedeutet das in der Praxis?
Wenn Sie noch nicht mit der Einführung von UDI angefangen haben, sollten Sie das Thema jetzt priorisieren. Die Verpflichtung zur UDI-Zuweisung für nicht-Legacy-Produkte gilt seit dem Geltungsbeginn der EU-MDR, dem 26. Mai 2021.
Die folgenden Schritte unterstützen Sie dabei, Ihr UDI-Projekt für MDSW umzusetzen:
1. Registrierung bei einer EU-akkreditierten Zuteilungsstelle
UDI-Codes müssen entsprechend den Regeln von einer EU-akkreditierten Zuteilungsstelle vergeben werden. Nach einem Ende 2018 veröffentlichten Aufruf zur Einreichung von Bewerbungen hat die Kommission gemäss Durchführungsbeschluss (EU) 2019/939 die folgenden Stellen benannt:
- GS1 AISBL
- Health Industry Business Communications Council (HIBCC)
- International Council for Commonality in Blood Banking Automation (ICCBBA)
- Informationsstelle für Arzneispezialitäten GmbH (IFA)
Die folgende Tabelle gibt einen Überblick über die Art der UDI-Aktivitäten und den geographischen Geltungsbereich für diese Zuteilungsstellen:
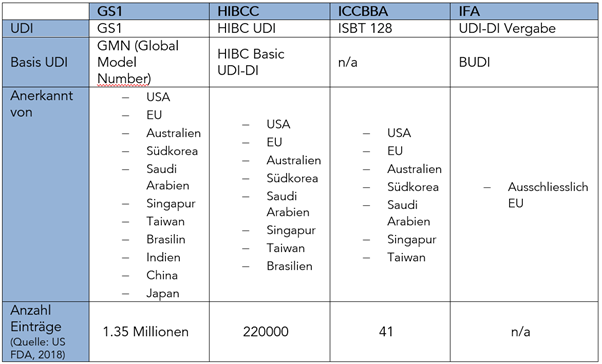
Die Art der UDI-Aktivitäten
Nach der Registrierung bei der Zuteilungsstelle Ihrer Wahl erhalten Sie ein Firmenpräfix, z.B. GS1 Global Company Prefix (GCP). Das ist der individuelle Identifikationsschlüssel, welcher Zugang zum System ermöglicht. Änderungen des rechtlichen Status der Firma (z.B. Namensänderungen, Fusionen, Übernahmen, Ausgliederungen) müssen der Zuteilungsstelle gemeldet werden.
2. Einrichten eines UDI-Management-Prozess
Das Qualitätsmanagementsystem des Herstellers sollte einen UDI-Management-Prozess enthalten, welcher mindestens die folgenden Aspekte abdeckt:
- Die Struktur der UDI-DI und UDI-PI des Herstellers
- Der Prozess und die Kriterien für die Gruppierung von MDSW unter einer BUDI
- Prozess für die UDI-PI-Kennzeichnung (sofern nicht im allgemeinen Produktkennzeichnungsverfahren beschrieben). Wenn die MDSW auf einem physischen Medium (z.B. CD, USB-Stick) geliefert wird, müssen ausserdem die IT- und Logistikaspekte von Barcodes gründlich durchdacht werden.
- UDI-DI und BUDI Change Management-Prozess, das heisst Kriterien, welche gemäss EU-MDR Anhang VI Teil C und MDCG 2019-5 einen neuen Code verlangen (sofern nicht im allgemeinen Change Management-Prozess beschrieben).
- Pflege des UDI-Datenstamm (Prozess zur Erstellung und Aufrechterhaltung der Liste aller BUDIs, UDI-DIs und deren zugeordneten zentralen Datenelementen). Die aktuelle Liste der UDI-DIs ist gemäss EU-MDR Artikel 27 (7) Teil der technischen Dokumentation der MDSW.
- Der Prozess zur elektronischen Übermittlung und Konsolidierung der UDI-Daten in EUDAMED und/oder in anderen spezifischen nationalen Datenbanken (zum Beispiel in der Schweiz).
Sofern Ihr Unternehmen noch keine Erfahrung mit GTIN Management hat, sollte dezidiert jemand mit regulatorischem und technischem Wissen benannt werden, der den UDI-Prozess während des gesamten Lebenszyklus des Produkts überwacht. Dies ist besonders relevant, wenn die MDSW auf einem physischen Medium (z.B. CD, USB-Stick) bereitgestellt wird und AIDC implementiert werden muss.
3. Zuweisung der einzelnen UDI-DIs
Für die im Handel als solche erhältliche MDSW muss der UDI-DI vom Hersteller auf der Ebene des Software-Systems zugewiesen werden. Bei Bereitstellung auf einem physischen Medium (z.B. CD oder USB-Stick) muss jeder Verpackungsebene (ausgenommen Versandbehälter) eine eigene UDI-DI zugewiesen werden. Die Global Trade Item Number (GTIN) von GS1 entspricht dem UDI-DI. Die Struktur des UDI-DI beinhaltet das Firmenpräfix, z.B. GS1 Global Company Prefix (GCP), der interne Code des Herstellers für das Produkt (z.B. die Modellnummer), und endet mit einer Prüfnummer, welche automatisch vom System berechnet wird.
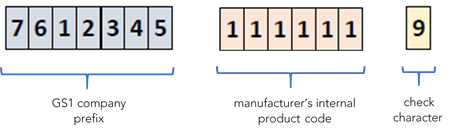
Beispiel einer UDI-DI-Struktur gemäss dem GS1 System
Bei der Vergabe/Aktualisierung aller Identifikationsnummern müssen EU-MDR Anhang VI, Teil C, Sektionen 3 und 6 berücksichtigt werden. Zudem müssen die relevanten Standards und Guidelines der jeweiligen UDI-Zuteilungsstelle befolgt werden.
4. Zuweisung der Basis UDI (BUDI)
Wenn Sie nur eine oder wenige MDSW Varianten haben, ist die Gruppierung der UDI-DIs auf Grundlage der BUDI-Kriterien (gleiche Risikoklasse, gleicher Verwendungszweck sowie die gleichen wesentlichen Design- und Herstellungsmerkmale) ein Kinderspiel. Beachten Sie aber, dass ein UDI-DI (GTIN) nicht als Ersatz für die Basis UDI-DI (GMN) genutzt werden darf. Die Gruppierung verschiedener MDS (verschiedene Risikoklasse oder Verwendungszweck oder Software-Architektur/Konfiguration) unter unterschiedlichen BUDIs muss sorgfältig durchdacht und dokumentiert werden, wobei auch Pläne für zukünftige MDSW Erweiterungen oder zusätzliche Funktionen berücksichtigt werden müssen. Wenn Ihre MDSW Produkte bereits unter der früheren Richtlinie 93/42/EWG über Medizinprodukte (MDD) CE-zertifiziert waren, haben Sie wahrscheinlich bereits festgestellt, dass die für das CE-Kennzeichen unter MDD verwendeten Medizinproduktefamilien/-gruppen möglicherweise keine geeigneten Produktgruppen gemäss BUDI-Kriterien bilden. Das Guidance-Dokument von MedTech Europe zu Basic UDI-DI Assignement enthält nützliche Tipps, wie Entscheidungen zur Gruppierung getroffen werden können.
Sobald Sie sich für die notwendigen BUDIs für die verschiedenen MDSW entschieden haben, können Sie die entsprechenden Codes erstellen. Ein vom Hersteller vergebener BUDI-Code darf aus maximal 25 Zeichen bestehen (die maximale Länge des UDI-DI wird von der Zuteilungsstelle festgelegt). Die Struktur des BUDI-Codes beinhaltet das Firmenpräfix, beispielsweise GS1 Global Company Prefix (GCP), und den internen Code für die Produktgruppe (numerisch oder alphanumerisch, Gross- und Kleinschreibung wird berücksichtigt) und ein zweistelliges Prüfzeichen basierend auf einem von der Zuteilungsstelle definierten Algorithmus.
Ein Beispiel für eine BUDI-Struktur gemäss dem GS1 System:
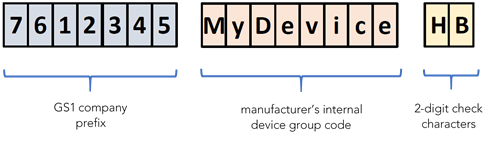
Beispiel einer Basis-UDI-Struktur (BUDI) basierend auf dem GS1 System
5. Erstellen der UDI-PIs
Die UDI-PIs sind numerische oder alphanumerische Codes, welche für die Kennzeichnung der Produktionseinheit des Produkts eingesetzt werden. Im Wesentlichen entsprechen Sie der Serien- oder Losnummer (abhängig vom Hersteller für einen bestimmten Medizinproduktetyp eingesetzten „Herstellungskontrollmechanismus “) mit zusätzlichen Informationen zu Produktions- oder Ablaufdaten. Sie erlauben damit eine eindeutige Identifizierung jeder Produkteinheit. Für die Generierung der UDI-PIs werden die Application Identifiers (AI) der jeweiligen Zuteilungsstelle verwendet. Als „Herstellungskontrollmechanismus“ gilt für MDSW die Software-Identifikation. Gemäss EU-MDR Anhang VI, Teil C, Abschnitt 6.5.1 ist sie Teil des UDI-PI. Das bedeutet, dass der AI für Serien- oder Losnummern für MDSW UDI-PI vor der Software Release Version verwendet werden würde. Bei einer Bereitstellung auf einem physischen Medium (z.B. CD oder USB-Stick) sind die UDI-PIs auch auf der Kennzeichnung erforderlich. Das folgende Beispiel zeigt den UDI-DI und UDI-PIs in einem 1D-Barcode (linear) und in einem 2D-Barcode (Datenmatrix), die beide mit einem Barcode-Scanner gelesen werden können sowie in einer von Menschen lesbaren Form (Verbindung der UDI-DI- und UDI-PIs-Teile, die entsprechenden AIs von GS1 sind vorangestellt).

Beispiel von UDI-DI und UDI-PIs gemäss den Barcodes (linear und Datenmatrix) von GS1.
Details über die AIs jeder einzelnen UDI-Zuteilungsstelle finden Sie in Anhang A des IMDRF Guides UDI WG application guide N48 FINAL: 2019.
6. Registrierung und Eingabe von UDI-Daten in EUDAMED
Vor der Produktregistrierung in EUDAMED muss der Hersteller (und jeder Produzent von Behandlungseinheiten oder Systemen) die Registrierung von Wirtschaftsakteuren im entsprechenden Modul der EUDAMED abschliessen und eine eigene einmalige Registrierungsnummer (SRN) gemäss EU-MDR erhalten. Eine detaillierte Beschreibung des Registrierungsprozesses findet sich auf der Webseite des entsprechenden Moduls. Die Registrierung ist seit Dezember 2020 möglich. Gemäss EU-MDR Artikel 123 (d) muss die Registrierung von Akteuren sechs Monate nach der vollständigen Funktionsfähigkeit der EUDAMED, die für den Mai 2022 geplant ist, abgeschlossen sein.
Die Rolle eines Unternehmens wird von der SRN eindeutig identifiziert. Die SRN wird von der zuständigen nationalen Behörde bei der Registrierungsanfrage des Unternehmens in EUDAMED vergeben. Die SRN-Struktur beinhaltet den ISO-Code des Landes, in welchem das Unternehmen seinen Sitz hat, die Abkürzung der Rolle des Wirtschaftsakteurs (beispielsweise „MF“ für manufacturer, „PR“ für Procedure Pack or System prodcuer, „AR“ for EU Authorized Representative oder „IM“ für EU importer), und eine 9-stellige Unternehmungskennung.
Ein Beispiel für eine SRN eines deutschen Herstellers:
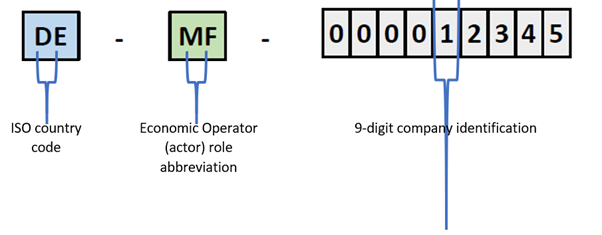
Beispiel einer einmaligen Registrierungsnummer (SRN) in EUDAMED.
Für Unternehmen, welche Ihren Sitz ausserhalb des EWR haben, ist der EU-Bevollmächtigte für die Überprüfung des Registrierungsantrags des Unternehmens vor der Einreichung bei der nationalen Behörde verantwortlich. Dies gilt ebenfalls für Schweizer Hersteller und Produzenten von Behandlungseinheiten und Systemen, da die Schweiz seit dem 26. Mai 2021 als Drittland gilt.
Ein entsprechendes Verfahren zur Vergabe von Schweizer Registrierungsnummern (CHRN) für Wirtschaftsakteure in der Schweiz wurde von der zuständigen Schweizer Behörde Swissmedic gemäss Artikel 55 der revidierten Schweizer Medizinprodukteverordnung MepV etabliert (MepV SR 812.213). Die Produktregistrierung im UDI-Modul der EUDAMED wird voraussichtlich im September 2021 möglich sein. Danach haben Hersteller bis im Mai 2024 Zeit, ihre Produkte (UDI-DI und BUDI) in EUDAMED gemäss EU-MDR Artikel 123 (e) zu registrieren, das heisst 18 Monate nach dem Datum in Artikel 123 (d).
Zum Zeitpunkt der Erstellung dieses Blogs steht immer noch nicht fest, wie dies in der Schweiz umgesetzt wird. Ein entsprechendes System zur Registrierung sollte von der Schweizer Behörde Swissmedic implementiert werden, um Informationen über UDIs über in der Schweiz in Verkehr gebrachte Produkte zu sammeln. Für eine Produktregistrierung in EUDAMED benötigen Sie den BUDI und sämtliche generierten UDI-DIs sowie die zugehörigen zentralen Datenelemente gemäss EU-MDR Anhang VI, Teil A. Das Guidance Dokument MDCG 2019-4 zu den Zeitplänen für die EUDAMED-Registrierung von Datenelementen enthält dazu weitere Informationen. Wichtig dabei ist, dass die vollständige Registrierung von Produkten eine Voraussetzung für die Meldung von schwerwiegenden Vorkommnissen oder Sicherheitskorrekturmassnahmen im Feld in EUDAMED bildet.
Nach der ersten Produktregistrierung in EUDAMED müssen Sie die UDI-Informationen in EUDAMED stets aktuell halten und neue Produkte (d.h. neue UDI-DI mit oder ohne neue BUDI) vor der Markteinführung registrieren. Jegliche Änderungen an zentralen Datenelementen, welche keine neue UDI-DI verlangen, müssen innert 30 Tagen nach der Änderung aktualisiert werden.
7. Falls erforderlich: Auswahl eines geeigneten UDI-Trägers
Die Verpflichtung zur Anbringung des UDI-Trägers auf der Produktkennzeichnung gemäß der EU-MDR erfolgt in Phasen gemäss dem folgenden Zeitplan:
|
Anforderung |
Klasse III und implantierbare Produkte |
Klasse IIb und Klasse IIa |
Klasse I |
|
Anbringen von UDI-Trägern auf Produktkennzeichnung gemäss EU-MDR Artikel 123(3)(f) |
26. Mai 2021 |
26. Mai 2023 |
26. Mai 2025 |
|
Direkte Kennzeichnung von wiederverwendbaren Produkten gemäss EU-MDR Artikel 123 (3)(g) |
26. Mai 2023 |
26. Mai 2025 |
26. Mai 2027 |
Die Kriterien zur Anbringung von UDI sind in EU-MDR Anhang VI, Teil C, Abschnitt 6.5.4 klar geregelt. Drei Situationen sind möglich:
- Bei auf einem physischen Medium gelieferter MDSW (z.B. CD, USB-Stick) muss jede Verpackungsebene sowohl die menschen- wie auch die maschinenlesbare (d.h. AIDC) Darstellung des kompletten UDI-PI tragen. Die UDI-PI auf dem physischen Medium mit der MDSW und seiner Verpackung muss mit der UDI-PI der Software auf Systemebene identisch sein.
- Bei nicht auf einem physischen Medium gelieferter MDSW, die aber eine Benutzeroberfläche hat (z.B. Mobile Apps, Webtools), ist nur die menschenlesbare Interpretation (HRI) des UDI-PI erforderlich. Sie muss auf einem für den Leser einfach zugänglichen Bildschirm in einem leicht lesbaren Klartextformat (z.B. in der „Über“ Datei, in Splashscreens oder auf dem Startbildschirm) bereitgestellt werden.
- Bei MDSW ohne Benutzeroberfläche (z.B. Middleware für Bildkonversionen) muss die menschenlesbare Darstellung der UDI-PI über eine Anwendungsprogrammierschnittstelle (API) bereitgestellt werden.
In jedem Falls muss das menschenlesbare Format der UDI-PI die Application Identifiers (AI) für den von der Zuteilungsstelle verwendeten Standard enthalten, um dem Benutzer die Identifizierung der UDI und die Bestimmung des zur Erstellung der UDI verwendeten Standards zu ermöglichen. Bei auf einem physischen Medium gelieferter MDSW muss der UDI-PI-Code auf jeder Verpackungsebene angebracht werden, von der Verwendungseinheit bis zur höchsten Verpackungsebene, ausgenommen sind logistische Einheiten. Es ist wichtig hervorzuheben, dass die Implementation eines UDI-PI-Trägers für ein Unternehmen ein komplexes Projekt darstellt, das nur erfolgreich durchgeführt werden kann, wenn es im Enterprise Resource Planning (ERP)-System des Unternehmens und mit dedizierten IT- und Logistikressourcen umgesetzt wird.
Die Kosten und der Zeitrahmen eines solchen Projekts müssen so früh wie möglich abgeschätzt werden und könnten zur Entscheidung führen, die MDSW nicht länger physisch, sondern online auszuliefern. Obwohl es sich dabei nicht um eine Anforderung der EU-MDR handelt, wird für den Fall, dass die MDSW auf einem physischen Medium geliefert wird und somit ein Barcode erforderlich ist, eine Überprüfung der Druckqualität des Barcodes empfohlen, um die Lesbarkeit in der gesamten Lieferkette zu gewährleisten. Die Standardreihe ISO 15415 bietet die für diesen Zweck relevanten Anforderungen für 2D- und lineare Barcodes.
8. UDIs während des gesamten Lebenszyklus der MDSW pflegen
Als Teil des Change Management-Prozesses müssen Änderungen der Software unter Berücksichtigung der Kriterien für die Erstellung einer neuen UDI-DI bewertet werden. Diese sind in EU-MDR Anhang VI, Teil C, Abschnitte 6.5.2 und 6.5.3 sowie im Guidance-Dokument MDCG 2018-5 zu UDI Assignment to Medical Device Software ausgeführt.
EU-MDR Anhang VI, Teil C, Abschnitt 6.5.2 verlangt eine neue UDI-DI für Software, „wenn Folgendes geändert wird:
(a) die ursprüngliche Leistung,
(b) die Sicherheit oder die bestimmungsgemäße Verwendung der Software,
(c) die Auswertung der Daten.
Zu diesen Änderungen gehören neue oder geänderte Algorithmen, Datenbankstrukturen, Betriebsplattformen und Architekturen oder neue Schnittstellen oder neue Kanäle für die Interoperabilität.“
Und gemäss EU-MDR Anhang VI, Teil C, Absatz 6.5.3 hängen geringfügige Änderungen der Software (welche nur eine neue UDI-PI und keine neue UDI-DI erfordern würden) „in der Regel mit Fehlerbehebungen, nicht Sicherheitszwecken dienenden Verbesserungen der Gebrauchstauglichkeit, Sicherheitspatches oder der Betriebseffizienz zusammen. Geringfügige Änderungen der Software werden mit einer herstellerspezifischen Kennzeichnungsart angegeben.“ Gemäss dem Guidance-Dokument MDCG 2018-1 ist eine neue UDI-DI immer dann erforderlich, wenn eine Änderung zu einer falschen Identifizierung des Geräts und/oder zu einer Mehrdeutigkeit in der Rückverfolgbarkeit führen. Das wird für MDSW im Guidance-Dokument MDCG 2018-5 weiter ausführt, das davon ausgeht, dass zur Gewährleistung der Rückverfolgbarkeit und korrekten Identifizierung der MDSW die folgenden Arten von Softwareänderungen eine neue UDI-DI verlangen würden:
- Jede Änderung der Basis UDI-DI
- Jede Änderungen, die sich auf die ursprüngliche Leistung, Sicherheit oder die Interpretation von Daten auswirkt.
- Eine Änderung des Namens oder der Handelsbezeichnung, der Versions- oder Modellnummer, wichtiger Warnhinweise und Kontraindikationen, der Sprache der Benutzeroberfläche
Wie sieht es mit Legacy-Produkten aus?
Legacy-Produkte, sogenannte Legacy Devices, d.h. Medizinprodukte, die nach dem Geltungsbeginn der EU-MDR weiterhin der Richtlinie 93/42/EWG (MDD) entsprechen, unterliegen nicht der UDI-Pflicht, sollten aber ebenfalls in EUDAMED registriert werden. Die EU-Kommission hat deshalb beschlossen, den Produkten anstelle einer Basis-UDI-DI eine EUDAMED-DI, und anstelle einer UDI-DI eine EUDAMED-ID zuzuweisen. Die EUDAMED-DI könnte entweder von EUDAMED generiert werden oder der Hersteller könnte den DI-Code teilweise zuweisen. Der EUDAMED-ID wird automatisch und vollständig von EUDAMED aus dem EUDAMED-DI-Code generiert. Die oben aufgezeigten Fristen für die EUDAMED-Registrierung gelten auch für Legacy-Produkte.
Weitere Informationen zu den operativen Aspekten für die Registrierung finden sich im Guidance-Dokument MDCG-2019-5 zu UDI for legacy devices. Alternativ empfehlen wir unseren Blogartikel zum Thema “Legacy Devices”.
Wie kann Decomplix Herstellern von medizinischer Software helfen?
Decomplix bietet für Hersteller Unterstützung für jeden Schritt des medizinischen CE-Zertifizierungsprozesses, einschliesslich medizinischer Software. Hier erfahren Sie mehr über unsere Dienstleistungen.



