
Risk Management for Medical Devices under EU MDR and ISO 14971
In this blog post, you will get an introduction to the medical device risk management requirements set forth by the EU MDR, as well as how the ISO 14971 standard can help you meet them.
Key takeaways:
- Risk management is required by law through the new Regulation (EU) 2017/745 on medical devices (EU MDR), to ensure that medical products are safe for patients, users and the environment
- Medical device manufacturers have to establish, implement, document and maintain a risk management system, which requires continuous maintenance and updates
- ISO 14971 standard defines the key requirements and steps in the process of medical device risk management
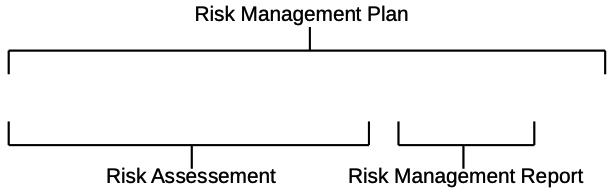
- To begin, you need to create a dedicated Risk Management Plan for your specific medical product.
Content
What is risk management for medical devices and why is it important?
Risk management is a fundamental part of a medical device’s development and throughout its entire lifecycle. Even though risk management is required by law through the new Regulation (EU) 2017/745 on medical devices (shortened as MDR), well executed risk management can also have a lot of benefits for your company.
By preventing Field Safety Corrective Actions (FSCA) of devices already placed on the market, it not only saves you costs and resources long term but also assures the wellbeing of your patients, users and the environment. The international standard ISO 14971 ‘Application of risk management to medical devices’ is essential for the implementation of these requirements. It describes a systematic risk management process and defines the evidence required.
To legally place medical devices on the market, authorities worldwide require that the products are subject to appropriate risk management. This is the only way to ensure the products are safe for patients, users and the environment. Since all possible risks are taken into account and are mitigated to the highest extent, the clinical benefits outweigh the residual risks.
Medical device risk management requirements of the MDR
Let’s start by having a look at the requirements of the MDR. Risk management is a basic requirement for medical device manufacturers and must be an integral part of the quality management system (MDR, Article 20 §9). The MDR further specifies that each medical device shall be designed and manufactured in a safe and effective way and that all risks are acceptable when weighed against their benefits to the patient.
It further stipulates that all risks need to be reduced as far as possible without adversely affecting the benefit-risk ratio. For that purpose, manufacturers have to establish, implement, document and maintain a risk management system which requires continuous and systematic updates (MDR, Annex I, Chapter I).

Even though risk management is required by law through the EU MDR, well executed risk management can also have a lot of benefits for your company.
Risk management process under ISO 14971
ISO 14971 is an internationally recognized standard. Currently applicable for the European Market under the MDR are the EN ISO 14971:2019 and the recently published Amendment 11 from 2021 (EN ISO 14971/A11:2021).
To understand the requirements of ISO 14971, it is important to familiarize yourself with the key terms. Essential is the difference between the terms harm, hazard and hazardous situation and risk.
Whereas a harm refers to the damage or injury to the health of an individual, property or environment, a hazard displays the potential source of that harm. The hazardous situation describes the circumstances in which the aforementioned individual, property or environment is/are exposed to one or more of the hazards. Lastly, risk is defined as the pure combination of the probability and harm of a specific hazardous situation.
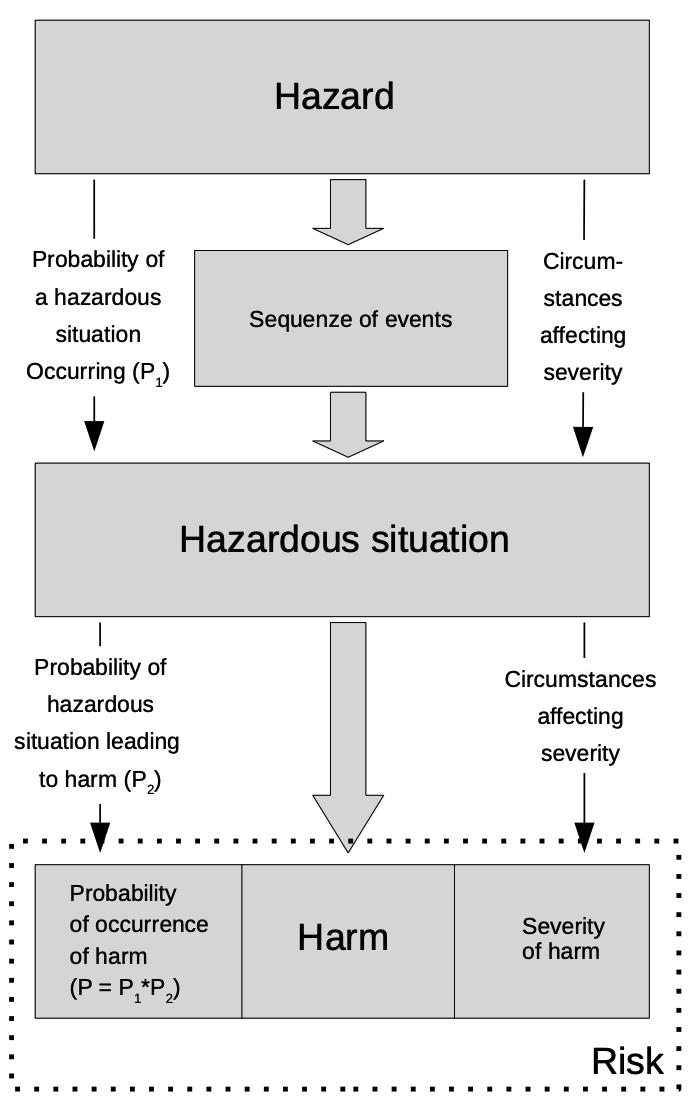
ISO 14971 defines the key differences between hazard, hazardous situation, harm and risk.
Another part of ISO 14971 that is key for good risk management are the numerous defined management responsibilities. For instance, a person from the management team is required to act as the person responsible for risk management.
In addition, it must be ensured that sufficient resources and sufficiently qualified personnel are made available. Furthermore, it is up to top management to define a policy defining risk acceptability criteria. Commonly, the policy for risk acceptability is described in the company’s quality management handbook.
The standard further specifies requirements for the risk management process. The figure below provides an overview of the recommended process steps.
Recommended process steps for medical device risk management.
How to create a Risk Management Plan
Product-specific risk management begins with the creation of a dedicated Risk Management Plan. It should address at least the following topics:
- General device description (including its intended use and classification)
- Methods for the evaluation of the overall residual risk and its acceptability
- Tasks and responsibilities assignment
- The scope of the risk management activities for all part of the device life cycle
- Requirements for the review of the performed activities
- Tasks for the verification of the implemented risk control measures
- Activities for the collection of production and post-production information
A: Risk Analysis
A risk analysis should be prepared for each medical device. Usually this is done in one or more documents called “Risk Analysis”. It is important to understand that this document shall cover the steps A to D for each identified risk. ISO/TR 24971 provides practical guidance on possible risk analysis techniques (e.g. Preliminary Hazard Analysis (PHA), Fault Tree Analysis (FTA), Failure Mode and Effects Analysis (FMEA), etc.).
The risk analysis part within the Risk Analysis document should contain at least:
- Identification of the device, various other points are to be generally addressed
- Intended use and reasonably foreseeable misuse
- Identification of the characteristics considered relevant for the safety of the device
- Identification of hazards and hazardous situations
A good starting point to identify the risks for a device is the risk screening questions from EN ISO 14971:2019 Annex C and ISO/TR 24971:2020 Annex A.
Based on this, you need to perform an estimate of the associated risks. The estimation of the identified risks shall incorporate the probability of occurrence of the harm and the severity of the harm.
B: Risk evaluation
All estimated risks must further be evaluated for their risk acceptability according to the risk acceptability defined in the Risk Management Plan.
It is important to notice that the MDR has additional requirements not present in EN ISO 14971:2019 that need to be considered as well. Firstly, all risks must be reduced as far as possible. For this reason, you must define risk control measures, even if the risk is acceptable, as long as there are control measures that actually reduce the risk.
Furthermore, MDR clearly states that risks are only acceptable when they are outweighed by the benefits. Therefore, you can only define your risk acceptability when you know the clinical benefits of your device. This means that you need the results from your Clinical Evaluation Report for the assessment of risk acceptability. Keep that also in mind for when you define your methods for the evaluation of the overall residual risk and its acceptability in the Risk Management Plan.
C: Risk control
For risks that are considered not acceptable, you have to determine appropriate measures to reduce these risks as far as possible. It is important to note that there are three different types of risk mitigations which shall be applied in the following order:
- Inherently safe design and manufacture
If the severity of a risk needs to be reduced, it needs to be made sure the device is physically incapable of creating the severity of an initially identified risk (e.g., change from 230V to 12V).
- Protective measures in the medical device itself or in the manufacturing process
If a certain design feature is indispensable, it needs to be made sure there are safety measures in place that keep resulting risks in check. Note that such control measures can only reduce the probability.
- Information for the user on the safe use
If there is a certain risk that cannot be eliminated by design or through safety measures (maybe because they enable the intended use in the first place), it needs to be made sure the user is properly instructed or trained, to reduce the probability of its occurring.
As part of the documentation, you must verify that all the planned control measures were implemented. Furthermore, it must be shown that the risk control measures actually reduced the risk they intended to reduce and whether they negatively impact existing risks or even create new ones. This can be done via internal verification of the risk control measures or through external verification (e.g. Basic Safety (IEC 60601 series), Biocompatibility (ISO 10993 series), Usability (EN 62366-1)).

Product-specific risk management begins with the creation of a dedicated Risk Management Plan.
D: Risk re-evaluation
After completion of the risk mitigation measures you have to again evaluate the risk acceptability according to the risk acceptability defined in the Risk Management Plan.
This evaluation should follow the same principles as the initial evaluation, based on methods for the evaluation of the overall residual risk and its acceptability in the Risk Management Plan. This means also applying the MDR requirements mentioned for the initial evaluation.
E: Risk management review
Before the medical device can be placed on the market, the proper execution of the Risk Management Plan needs to be reviewed. The results of this evaluation need to be documented in the Risk Management Report.
Normally, the plan is updated only if something fundamental changes. This means that if there were any deviations from the plan, make sure to document them in the report. Consider this before changing the plan, oftentimes it does not need to be changed immediately.
The review further has to state whether the overall residual risks are acceptable. Lastly, the report also needs to assess if there are appropriate methods in place for the collection and evaluation of information from the production and post-production phase.
F: Production and post-production activities
As indicated above, you have to implement processes and methods for the collection and review of production and post-production data (i.e., after the device has been placed on the market). The information must be collected throughout the whole life-cycle of the device. Relevant data may be feedback from users, information generated by the supply chain, publicly available information, etc.
All of the above-mentioned data needs to be reviewed in order to possibly identify previously unrecognized hazards or hazardous situations, changes to risk acceptability based on increased severity or probability, changes to the state-of-the-art and therefore changed global risk acceptability. If the data review shows that actions are needed, this must be documented. The actions (e.g. FSCA) need to be planned and recorded.
In conclusion – what you need to know about risk management
Risk management and its documentation is a lively process and requires continuous maintenance and update. Make sure to define appropriate resources to update your risk management in regular intervals and to closely monitor any trends or indications regarding the safety of your medical device.
Within this blog post, we have seen that risk management is a fundamental tool in the development, manufacture and continuous improvement of medical devices. It is mandatory for every medical device and indispensable for patient and user safety. The international standard ISO 14971 specifies the necessary process steps and documentation.
Nevertheless, there are some requirements from EU MDR not covered by ISO 14971, but they must be considered. After all, ISO 14971 and ISO/TR 24971 are very helpful tools to better understand how to implement compliant risk management for your device. With Commission Implementing Decision (EU) 2022/757 of 11 May 2022, which was published in the Official Journal on 17 May 2022, EN ISO 14971:2019 is now harmonized with the MDR.
This means EN ISO 14971:2019 and EN ISO 14971:2019/A11:2021 are applicable for the presumption of conformity to the requirements of the MDR. The Annex Z explains the link between the clauses in the standards and the GSPRs of the MDR with which they are linked.
How Decomplix can help
Are there specific questions you would like to discuss? As you now know, the risk management is very specific with the device and its intended use. We are happy to support in you in setting up your compliant risk management. You can learn more about our services here.



