
Risikomanagement für Medizinprodukte unter EU-MDR und ISO 14971
In diesem Blogbeitrag erhalten Sie eine Einführung in die Anforderungen an das Risikomanagement von Medizinprodukten, die in der EU-MDR festgelegt sind, und erfahren, wie die Norm ISO 14971 Ihnen helfen kann, diese zu erfüllen.
Die wichtigsten Erkenntnisse:
- Risikomanagement ist durch die europäische Verordnung (EU) 2017/745 über Medizinprodukte (MDR) gesetzlich vorgeschrieben, um sicherzustellen, dass Medizinprodukte für Patienten, Anwenderinnen und die Umwelt sicher sind.
- Hersteller von Medizinprodukten müssen ein Risikomanagementsystem einrichten, umsetzen, dokumentieren und aufrechterhalten, das kontinuierlich gepflegt und aktualisiert werden muss.
- Die Norm ISO 14971 definiert die wichtigsten Anforderungen und Schritte im Prozess des Risikomanagements für Medizinprodukte.
- Zu Beginn müssen Sie einen spezifischen Risikomanagementplan für Ihr Medizinprodukt erstellen.
Inhalt:
Was ist Risikomanagement und warum ist es wichtig?
Das Risikomanagement ist ein elementarer Teil der Entwicklung und des gesamten Lebenszyklus eines Medizinproduktes. Auch wenn die Europäische Verordnung über Medizinprodukte (EU) 2017/745 (kurz MDR) das Risikomanagement gesetzlich vorschreibt, kann ein gut geführtes Risikomanagement auch sonst viele Vorteile für Ihr Unternehmen haben.
Durch das Vermeiden von Sicherheitskorrekturmassnahmen im Feld (Field Safety Corrective Actions FSCA) für bereits in Verkehr gebrachte Produkte, können Sie langfristig Kosten sparen und Ressourcen schonen, während zudem das Wohl Ihrer Patienten, Anwender und der Umwelt gewährleistet wird. Der internationale Standard ISO 14971 «Application of risk management to medical devices” ist für die Implementierung dieser Anforderungen grundlegend. Er beschreibt einen systematischen Risikomanagementprozess und definiert den erforderlichen Nachweis.
Um rechtlich ein Medizinprodukt in Verkehr zu bringen, schreiben Behörden weltweit vor, dass die Produkte einem angemessenen Risikomanagement unterzogen werden. Dies ist der einzige Weg um sicherzustellen, dass die Produkte für Patienten, Anwender und die Umwelt sicher sind. Da alle möglichen Risiken geprüft und in höchstem Mass gemindert werden, überwiegen die klinischen Vorteile das Restrisiko.
Anforderungen der MDR an das Risikomanagement von Medizinprodukten
Beginnen wir mit einem Blick auf die Anforderungen der MDR. Das Risikomanagement ist eine grundlegende Anforderung für Medizinproduktehersteller und muss ein integrativer Teil des Qualitätsmanagementsystems sein (MDR, Artikel 20 §9). Die MDR spezifiziert zudem auch, dass jedes Medizinprodukt auf eine sichere und wirksame Art entworfen und hergestellt werden soll und dass alle Risiken vertretbar sind, wenn sie am Nutzen für den Patienten gemessen werden.
Sie regelt zudem, dass alle Risiken so weit wie möglich gemindert werden müssen, ohne das Nutzen-Risiko-Verhältnis negativ zu beeinflussen. Zu diesem Zweck haben die Hersteller ein Risikomanagementsystem, das eine regelmässige und systematische Aktualisierung erfordert, zu etablieren, implementieren, dokumentieren und zu unterhalten (MDR, Anhang I, Kapitel I).

Abgesehen davon, dass die EU-MDR das Risikomanagement gesetzlich vorschreibt, kann ein gut geführtes Risikomanagement auch sonst viele Vorteile für Ihr Unternehmen haben.
Risikomanagementprozess nach ISO 14971
ISO 14971 ist ein international anerkannter Standard. Gegenwärtig für den Europäischen Markt unter der MDR anwendbar sind EN ISO 14971:2019 und die kürzlich veröffentliche Änderung 11 von 2021 (EN ISO 14971/A11:2021).
Um die Anforderung von ISO 14971 zu verstehen, ist es wichtig, dass Sie sich mit den Schlüsselbegriffen vertraut machen. Grundlegend ist der Unterschied zwischen den Begriffen Schaden, Gefährdung, gefährliche Situation und Risiko.
Während Schaden sich auf Beeinträchtigung oder Verletzung der Gesundheit einer Person, von Eigentum oder der Umwelt bezieht, zeigt Gefährdung die mögliche Quelle eines solchen Schadens auf. Die gefährliche Situation beschreibt die Umstände, in denen die oben genannte Person, Eigentum oder die Umwelt einer oder mehreren Gefährdungen ausgesetzt ist/sind. Schliesslich wird Risiko als pure Kombination der Möglichkeit und Gefährdung einer spezifischen gefährlichen Situation beschrieben.
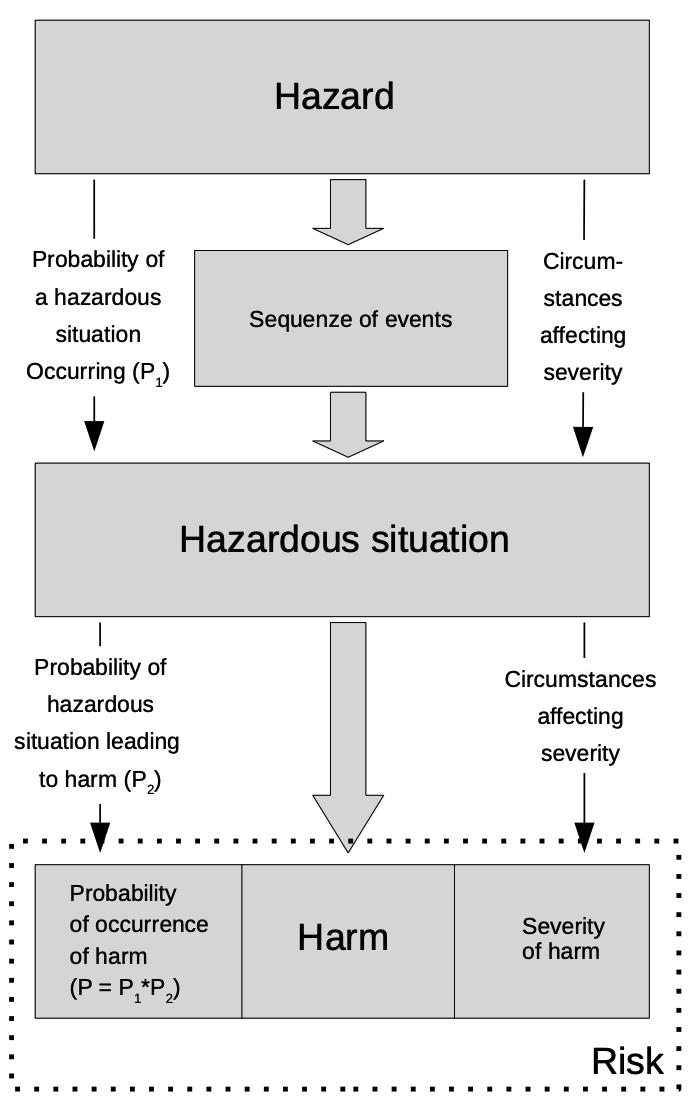
ISO 14971 definiert die wichtigsten Unterschiede zwischen Gefährdung, gefährlicher Situation, Schaden und Risiko.
Ein weiterer Teil von ISO 14971, der für gutes Risikomanagement grundlegend ist, sind die verschiedenen definierten Managementverantwortlichkeiten. Zum Beispiel muss eine Person vom Managementteam als Verantwortliche/r für das Risikomanagement amtieren.
Zudem muss sichergestellt werden, dass ausreichend Ressourcen und qualifizierte Mitarbeitende zur Verfügung gestellt werden. Auch liegt die Bereitstellung einer Richtlinie, die Risikoakzeptanzkriterien definiert, in der Verantwortung des oberen Managements. Üblicherweise ist die Richtlinie für die Risikoakzeptanz Teil des Qualitätsmanagementhandbuchs des Unternehmens.
Der Standard spezifiziert weiter Anforderungen für den Risikomanagementprozess. Die unten folgende Darstellung zeigt eine Übersicht über die empfohlenen Prozessschritte.
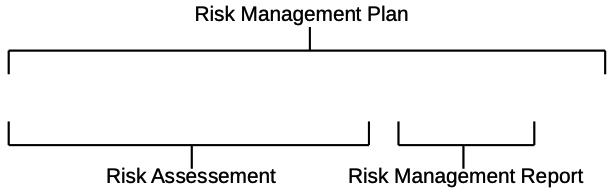
Empfohlene Prozessschritte für das Risikomanagement von Medizinprodukten.
Wie man einen Risikomanagementplan erstellt
Das produktspezifische Risikomanagement beginnt mit der Erstellung eines Risikomanagementplans. Dieser sollte mindestens die folgenden Themen beinhalten:
- Allgemeinte Produktbeschreibung (einschliesslich der Zweckbestimmung und Klassifizierung)
- Methoden zur Bewertung des verbundenen Restrisikos und dessen Vertretbarkeit
- Zuweisung von Aufgaben und Verantwortlichkeiten
- Geltungsbereich der Tätigkeiten für das Risikomanagement für alle Teile des Lebenszyklus des Medizinproduktes
- Anforderungen an die Überprüfung der durchgeführten Tätigkeiten
- Aufgaben zur Verifizierung der implementierten Risikokontroll-Massnahmen
- Tätigkeiten zur Sammlung von Informationen während der Herstellung und in der der Herstellung nachgelagerten Phase
A: Risikoanalyse
Es sollte für jedes Medizinprodukt eine Risikoanalyse durchgeführt werden. Normalerweise wird dies anhand eines oder mehrerer Dokumente gemacht. Es ist wichtig zu verstehen, dass dieses Dokument die Schritte A bis D für jedes identifizierte Risiko abdecken soll. ISO/TR 24971 bietet praktische Anleitung für mögliche Risikoanalyse-Techniken (z. B. vorläufige Gefahrenanalyse, Fehlerbaumanalyse, Auswirkungsanalyse, etc.)
Der Risikoanalyseteil der Risikoanalyse-Dokumentation sollte mindestens Folgendes beinhalten:
- Identifikation des Medizinprodukts, verschiedene andere Punkte müssen allgemein adressiert werden
- Zweckbestimmung und vernünftigerweise vorhersehbare Fehlanwendungen
- Identifikation der für die Sicherheit des Medizinproduktes berücksichtigten Charakteristiken
- Identifikation von Gefährdungen und gefährlichen Situationen
Ein guter Ausgangspunkt zur Risikoidentifikation für ein Medizinprodukt sind die Risikoscreening-Fragen aus EN ISO 14971:2019 Anhang C und ISO/TR 24971:2020 Anhang A.
Darauf basierend müssen Sie die assoziierten Risiken abschätzen. Die Abschätzung der identifizierten Risiken sollte die Auftretenswahrscheinlichkeit des Schadens und den Schweregrad des Schadens miteinbeziehen.
B: Risikobewertung
Alle abgeschätzten Risiken müssen anhand der im Risikoplan definierten Risikoakzeptanz weiter evaluiert werden.
Es ist auch wichtig zu wissen, dass die MDR zusätzliche Anforderungen festlegt, die nicht in der EN ISO 14971:2019 enthalten sind, und die ebenfalls zu berücksichtigen sind. Aus diesem Grund sollten Sie Risikokontroll-Massnahmen definieren, auch wenn das Risiko vertretbar ist, so lange die Kontrollmassnahmen das Risiko effektiv mindern.
Zudem sagt die MDR klar, dass Risiken nur vertretbar sind, wenn sie von den Vorteilen überwogen werden. Daher können Sie Ihre Risikoakzeptanz nur definieren, wenn Sie die klinischen Nutzen Ihres Medizinprodukts kennen. Das bedeutet, dass Sie die Ergebnisse Ihres Berichts über die klinische Bewertung benötigen, um die Bewertung der Risikoakzeptanz durchzuführen. Berücksichtigen Sie dies auch, wenn Sie Ihre Methoden für die Bewertung der verbundenen Restrisiken und deren Vertretbarkeit im Risikomanagementplan definieren.
C: Risikokontrolle
Für die als nicht vertretbar bewerteten Risiken müssen Sie angemessene Massnahmen bestimmen, welche diese Risiken so weit wie möglich minimieren. Es ist wichtig zur Kenntnis zu nehmen, dass es drei verschiedene Arten zur Risikominderung gibt, die in der nachfolgenden Reihenfolge angewendet werden sollen:
- Inhärent sichere Auslegung und Herstellung
Wenn der Schweregrad eines Risikos gemindert werden muss, soll sichergestellt werden, dass das Medizinprodukt physisch den Schweregrad des ursprünglich identifizierten Risikos unmöglich erzeugen kann (z.B. Wechsel von 230V zu 12V).
- Schutzmassnahmen im Medizinprodukt selbst oder im Herstellprozess
Falls bestimmte Designmerkmale unentbehrlich sind, muss sichergestellt werden, dass Sicherheitsmassnahmen vorhanden sind, um die daraus entstehenden Risiken zu kontrollieren. Berücksichtigen Sie, dass solche Kontrollmassnahmen lediglich die Wahrscheinlichkeit reduzieren.
- Informationen für den Anwender zur sicheren Verwendung
Falls ein bestimmtes Risiko vorhanden ist, das nicht durch Design oder Sicherheitsmassnahmen eliminiert werden kann (vielleicht weil es den Verwendungszweck erst ermöglicht), muss sichergestellt werden, dass der Anwender richtig instruiert oder geschult wird, um die Wahrscheinlichkeit des Auftretens zu reduzieren.
Als Teil der Dokumentation müssen Sie verifizieren, dass alle geplanten Kontrollmassnahmen implementiert wurden. Darüber hinaus muss gezeigt werden, dass die Risikokontroll-Massnahmen das Risiko, das sie vermindern sollen, tatsächlich reduzieren. Auch muss gezeigt werden, ob sie bestehende Risiken negativ beeinflussen oder gar neue schaffen. Dies kann über die interne Verifizierung der Risikokontrollmassnahmen erreicht werden oder durch die externe Verifizierung (z.B. Basic Safety (IEC 60601 Serien), Biokompatibilität (ISO 10993 Serien), Usability (EN 62366-1)).

Das produktspezifische Risikomanagement beginnt mit der Erstellung eines Risikomanagementplans.
D: Neubewertung von Risiken
Nach Abschluss der Risikominderungsmassnahmen sollten Sie die Vertretbarkeit der Risiken anhand der im Risikomanagementplan definierten Risikoakzeptanz erneut bewerten.
Diese Evaluation sollte die gleichen Prinzipien wie die ursprüngliche Evaluation verwenden, basierend auf den Methoden für die Bewertung des verbundenen Restrisikos und dessen Vertretbarkeit.
Das bedeutet, dass auch die erwähnten MDR-Anforderungen in der ursprünglichen Evaluation verwendet werden sollten.
E: Risikomanagement-Review
Bevor das Medizinprodukt in Verkehr gebracht werden kann, muss die korrekte Ausführung des Risikomanagementplans geprüft werden. Das Ergebnis dieser Evaluation muss im Risikomanagementbericht dokumentiert werden.
Normalerweise wird der Plan nur aktualisiert, wenn etwas grundlegend ändert. Das bedeutet, dass Sie im Fall von Abweichungen vom Plan sicherstellen sollten, dass diese im Bericht dokumentiert werden. Berücksichtigen Sie dies auch, bevor Sie den Plan verändern. Oftmals muss er nicht sofort geändert werden.
Das Review muss zudem auch aussagen, ob die verbundenen Restrisiken vertretbar sind. Schliesslich muss der Bericht auch bewerten, ob angemessene Methoden für das Sammeln und die Bewertung von Informationen während der Herstellung sowie in der der Herstellung nachgelagerten Phase vorhanden sind.
F: Tätigkeiten während der Herstellung und in der der Herstellung nachgelagerten Phase
Wie oben erwähnt, sollten Sie Prozesse und Methoden für das Sammeln und die Prüfung von Daten während der Herstellung und in der der Herstellung nachgelagerten Phase (i.e. nachdem das Medizinprodukt in Verkehr gebracht worden ist) implementieren. Die Informationen müssen während des gesamten Lebenszyklus des Medizinprodukts gesammelt werden. Relevante Daten können Rückmeldungen von Anwendern, in der Lieferkette generierte Informationen, öffentlich vorhandenen Informationen, etc. sein.
Alle der oben erwähnten Daten müssen geprüft werden, um mögliche bislang unerkannte Gefährdungen oder gefährliche Situationen, Veränderungen hinsichtlich der Vertretbarkeit von Risiken aufgrund eines grösseren Schweregrads oder einer höheren Wahrscheinlichkeit, Änderungen am Stand der Technik und einer dadurch veränderten globalen Risikoakzeptanz identifizieren zu können. Wenn die Prüfung der Daten zeigt, dass Tätigkeiten erforderlich sind, muss dies dokumentiert werden. Die Tätigkeiten (z.B. FSCA) müssen geplant und dokumentiert werden.
Zusammenfassend – was Sie über Risikomanagement wissen müssen
Das Risikomanagement und dessen Dokumentation ist ein lebendiger Prozess, der laufend gepflegt und aktualisiert werden muss. Stellen Sie sicher, dass Sie angemessene Ressourcen definieren, um Ihr Risikomanagement in regelmässigen Abständen zu aktualisieren und mögliche Trends oder Indikationen hinsichtlich Ihres Medizinprodukts genau zu verfolgen.
In diesem Beitrag haben wir gesehen, dass das Risikomanagement ein fundamentales Werkzeug in der Entwicklung, Herstellung und der laufenden Verbesserung von Medizinprodukten ist. Es ist für jedes Medizinprodukt vorgeschrieben und für die Sicherheit der Patienten und Anwender unentbehrlich. Der internationale Standard ISO 14971 spezifiziert die notwendigen Prozessschritte und die Dokumentation.
Dennoch gibt es einige Anforderungen der EU-MDR, die von der ISO 14971 nicht abgedeckt werden, die aber berücksichtigt werden müssen. Schließlich sind ISO 14971 und ISO/TR 24971 sehr hilfreiche Instrumente, um besser zu verstehen, wie man ein konformes Risikomanagement für sein Produkt implementiert. Mit dem Durchführungsbeschluss (EU) 2022/757 der Kommission vom 11. Mai 2022, der am 17. Mai 2022 im Amtsblatt veröffentlicht wurde, ist die EN ISO 14971:2019 nun mit der MDR harmonisiert.
Das bedeutet, dass EN ISO 14971:2019 und EN ISO 14971:2019/A11:2021 für die Vermutung der Konformität mit den Anforderungen der MDR anwendbar sind. Der Anhang Z erläutert den Zusammenhang zwischen den Abschnitten in den Normen und den GSPR der MDR, mit denen sie verknüpft sind.
Wie Decomplix helfen kann
Haben Sie spezifische Fragen, die Sie gerne besprechen möchten? Wie Sie nun Wissen, ist das Risikomanagement hinsichtlich des Medizinprodukts und der Zweckbestimmung sehr spezifisch. Wir unterstützen Sie gerne mit dem Aufbau Ihres konformen Risikomanagements. Sie können hier mehr über unsere Dienstleistungen erfahren.



