Medizinproduktesoftware (MDSW) gemäss EU-MDR und IVDR
Ist Ihre Software ein Medizinprodukt? Wenn Sie unsicher sind, lesen Sie weiter. Entwickeln Sie Software, die für medizinische Zwecke verwendet wird? Wenn dem so ist, und Sie noch nicht mit der EU-MDR und IVDR vertraut sind, erfahren Sie hier mehr über die CE-Kennzeichnung von Medizinproduktesoftware (MDSW).
Ersetzt die Version vom 02.03.2019
Wichtige Erkenntnisse
- Ein medizinischer Zweck ist der entscheidende Faktor, ob eine Software als MDSW gilt. Auch Zwecke wie die “Vorhersage” und “Prognose” führen zu einer MDSW. Es gilt also, genau hinzuschauen, um die Qualifizierung als MDSW wenn möglich zu vermeiden.
- Die Klassifizierung von Software hat sich im Rahmen der EU-MDR und der IVDR drastisch geändert. Es fällt praktisch keine MDSW in eine tiefe Risikoklasse, weshalb meistens eine Benannte Stelle in den CE-Kennzeichnungsprozess eingebunden werden muss. Überprüfen Sie die Klassifizierungsregeln, um Fehleinstufungen und Verzögerungen bei der Markteinführung zu vermeiden.
- Der “Legacy”-Status kann auch für MDSW gelten. Dieser Status ist aber mit strengen Einschränkungen verbunden. Vergewissern Sie sich, dass Sie die wesentlichen Änderungen kennen, die zum Verlust des “Legacy”-Status führen.
Inhalt
Was ist Medizinproduktesoftware (MDSW)?
Zusammenfassend lässt sich sagen, dass die folgenden drei Bedingungen erfüllt sein müssen, damit eine Software als MDSW eingestuft wird:
- Sie muss einen eigenen medizinischen Zweck haben,
- Sie führt eine Aktion mit Daten durch, die über Speicherung, Archivierung, Kommunikation, einfache Suche oder verlustfreie Kompression hinausgeht, und
- Sie ist für den Nutzen einzelner Patienten bestimmt.
MDSW ist eine Software, die (allein oder in Kombination) für einen der Zwecke verwendet wird, die in der Definition von “Medizinprodukt” in Art. 2 Abs.1 der Verordnung (EU) Nr. 2017/745 (EU-MDR) oder der Verordnung (EU) Nr. 2017/746 (IVDR) beschriebenen sind.
Die rechtliche Definition des Begriffs “Medizinprodukt” umfasst die folgenden Zwecke:
- Diagnose, Prävention, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung oder Kompensation einer Verletzung oder Behinderung,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Prozesses oder Zustands,
- Bereitstellung von Informationen durch In-vitro-Untersuchung von aus dem menschlichen Körper stammenden Proben, einschliesslich Organ-, Blut- und Gewebespenden,
- Kontrolle oder Unterstützung der Empfängnis,
- Reinigung, Desinfektion oder Sterilisation von Medizinprodukten.
Die EU-MDR hat eine kleine, aber entscheidende Erweiterung der Definition des Medizinprodukts eingeführt, nämlich die beiden Zwecke “Vorhersage” und “Prognose” einer Krankheit. Software, die nach der bisherigen Richtlinie nicht als Medizinprodukt eingestuft wurde, könnte wegen diesen neuen Zwecken nun unter die Definition eines Medizinprodukts fallen.
Kurz gesagt: eine MDSW ist eine Software, die eigenständig einen Zweck eines Medizinprodukts hat.
Die Zweckbestimmung ist denn auch ein entscheidender Faktor bei der Einstufung als MDSW. Die Tatsache, dass eine Software im Gesundheitswesen eingesetzt wird, macht sie noch nicht zur MDSW.
Relevant ist die Art der Datenverarbeitung. Laut MDCG 2019-11 gilt Software nicht als MDSW, wenn sich die Datenverarbeitung auf folgendes beschränkt:
- Speicherung,
- Archivierung,
- Kommunikation, verstanden als “the flow of information from one point, known as the source, to another, the receiver”
- einfache Suche, beschrieben als “the retrieval of records by matching record metadata against record search criteria or to the retrieval of information […] (e.g. library functions)”, oder
- verlustfreie Komprimierung, d.h. die Verwendung eines Komprimierungsverfahrens, das die exakte Rekonstruktion der ursprünglichen Daten ermöglicht.
Wenn die Software hingegen medizinische Informationen verarbeitet, analysiert, erstellt oder verändert, kann sie als MDSW eingestuft werden, wenn sich diese Vorgänge aus einer medizinischen Zweckbestimmung ergeben.
«Kurz gesagt: eine MDSW ist eine Software, die eigenständig einen Zweck eines Medizinprodukts hat.»
Ist die Datenverarbeitung jedoch nicht zum Nutzen einzelner Patienten bestimmt, so gilt die Software wiederum nicht als MDSW. Beispielsweise Software, die Bevölkerungsdaten zusammenfasst, allgemeine Diagnose- oder Behandlungspfade bereitstellt, wissenschaftliche Literatur (z.B. medizinische Atlanten) oder Software, die nur für epidemiologische Studien oder Register bestimmt ist. Dies steht im Einklang mit Erwägungsgrund 19 der EU-MDR:
“… während Software für allgemeine Zwecke, auch wenn sie in Einrichtungen des Gesundheitswesens eingesetzt wird, sowie Software, die für Zwecke in den Bereichen Lebensstil und Wohlbefinden eingesetzt wird, kein Medizinprodukt ist.”
Ein Softwarehersteller kann also eine MDSW-Einstufung vermeiden, indem er eine dieser drei Bedingungen nicht erfüllt.
Es ist auch möglich, die Softwarefunktionalität, die als MDSW eingestuft wird, von der nicht als MDSW eingestuften zu trennen. Und zwar durch klar abgegrenzte Module in der Softwarearchitektur. Weitere Einzelheiten zu einem modularen Ansatz für MDSW finden Sie unter Ist die MDSW-Modularisierung ein geeigneter Ansatz?
Die folgenden Arten von Software gelten auf Grund ihrer Zweckbestimmung nicht als MDSW, unterliegen aber dennoch den Anforderungen der EU-MDR bzw. IVDR:
- Software, die der rechtlichen Definition von “Zubehör” zu einem Medizinprodukt oder einem In-vitro-Diagnostikum (IVD) entspricht,
- Software, die Produkten ohne medizinische Zweckbestimmung entspricht, wie in Anhang XVI der EU-MDR aufgeführt,
- Software, die die Verwendung eines (Hardware-)Medizinprodukts oder IVD steuert oder beeinflusst, und dabei keine eigene medizinische Zweckbestimmung hat und keine eigenen Informationen erzeugt.
Eine MDSW ist eine Software, die eigenständig einen Zweck eines Medizinprodukts hat.
Wann ist ein MDSW ein In-vitro-Diagnostikum (IVD)?
Software, die als MDSW eingestuft wird, kann unter die EU-MDR oder die IVDR fallen. Welche Verordnung gilt, wird einfach anhand der Daten vorgenommen, die von der MDSW verarbeitet werden. MDSW unterliegt also der IVDR, wenn:
- sie Informationen bereitstellt, die ausschliesslich auf mit IVDs gewonnenen Daten basieren, oder
- ihre Zweckbestimmung im Wesentlichen von IVD-Datenquellen bestimmt wird.
So sind z.B. Krankheitsrisikorechner, bei denen die Eingaben hauptsächlich aus Blut- und/oder Urintestergebnissen stammen, MDSW gemäss der IVDR (“IVDR MDSW”). Umgekehrt sind Krankheitsrisikorechner, deren Eingaben aus Elektrokardiogrammen oder Bildern stammen, MDSW gemäss der EU-MDR (“EU-MDR MDSW”).
Software, die lediglich die Darstellung von IVD-Ergebnissen verändern soll, gilt nicht als MDSW. Die MDCG 2019-11 enthält folgende Funktionsbeispiele, die als “Änderung der Darstellung” angesehen werden:
- Grundlegende arithmetische Operationen (z.B. Mittelwert, Umrechnung von Einheiten),
- die Darstellung der Ergebnisse in Abhängigkeit von der Zeit,
- ein Vergleich des Ergebnisses mit den vom Benutzer festgelegten Akzeptanzgrenzen.
Und wenn die Software notwendig ist und ausschliesslich dazu dient, die Rohdaten eines IVDs für den Benutzer lesbar zu machen, wäre dies eine MDSW, die das entsprechende IVD steuert oder beeinflusst.
Was ist der Unterschied zwischen MDSW und Software als Medizinprodukt?
Software als Medizinprodukt (SaMD für «software as medical device») ist ein internationaler Begriff, der von der IMDRF definiert wurde und in die Gesetzgebung vieler Länder (einschliesslich des Vereinigten Königreichs) übernommen wurde, während MDSW ausschliesslich ein EU-Begriff ist.
Die IMDRF/SaMD WG/N10FINAL2013 definiert SaMD wie folgt:
“Software intended to be used for one or more medical purposes that perform the purposes without being part of a hardware medical device. Software is a SaMD regardless of the fact that it needs hardware to execute. Regardless of whether it operates on general-purpose IT equipment, in “the cloud” or on the computing platform of a [hardware] of a medical device, as long as it is not “necessary for the [hardware] medical device to achieve its intended medical purpose.”
SaMD betrifft nur unabhängige-Software, und schliesst jegliche Software, die in ein physisches Medizinprodukt “eingebettet” ist, nicht mit ein. Hingegen kann MDSW sowohl unabhängig als auch Teil eines Hardware-Medizinprodukts sein, wenn sie neben dem Zweck der Steuerung des Hardware-Medizinprodukts auch einen eigenen medizinischen Zweck hat. Einige Beispiele für “eingebettete” Software mit eigenem medizinischen Zweck sind in der MDCG 2019-11 aufgeführt:
- Software zur Melanom-Bildanalyse, die einen Nahinfrarot-Laserscanner ansteuern soll.
- MDSW zur Messung und Übertragung des Blutzuckerspiegels und zur Berechnung der erforderlichen Insulindosis.
Beachten Sie auch, dass der Begriff “standalone”-Software, der in der Medizintechnikbranche weit verbreitet ist, kein Synonym für “unabhängige” MDSW ist. Eine “standalone” MDSW kann immer noch ein medizinisches Hardware-Gerät steuern oder beeinflussen.
Wird Software, die in einer Gesundheitseinrichtung entwickelt und verwendet wird, als MDSW betrachtet?
Unter Umständen.
Wenn diese Software einen eigenständigen Zweck als Medizinprodukt hat, zum Nutzen einzelner Patienten bestimmt ist und Daten auf eine Art und Weise verarbeitet, die nicht nur der Speicherung, Archivierung, Kommunikation, verlustfreien Komprimierung oder einfachen Suche dient, kann sie als MDSW eingestuft werden.
«MDSW, die ausschliesslich in einer Gesundheitseinrichtung hergestellt und verwendet werden, müssen nicht mit dem CE-Kennzeichen versehen werden.»
Jedes Produkt (einschliesslich MDSW), das in einer Gesundheitseinrichtung hergestellt und nur in dieser Einrichtung verwendet wird, gilt als in Betrieb genommen – gemäss Art. 5 Abs. 4 sowohl der EU-MDR als auch der IVDR. Für solche Produkte gelten die in Art. 5 Abs. 5 der EU-MDR bzw. IVDR genannten Voraussetzungen, die jedoch weit weniger umfangreich sind als die Anforderungen, die für in Verkehr gebrachte Produkte gelten.
MDSW, die ausschliesslich in einer Gesundheitseinrichtung hergestellt und verwendet werden, müssen nicht mit dem CE-Kennzeichen versehen werden. Dennoch müssen sie die folgenden Anforderungen erfüllen:
- Sie müssen im Rahmen eines geeigneten Qualitätsmanagementsystems entwickelt/produziert und verwendet werden.
- Sie müssen über eine von der Gesundheitseinrichtung ausgestellte Begründung verfügen, aus der hervorgeht, dass die spezifischen Bedürfnisse der Patienten-Zielgruppe nicht durch ein gleichwertiges Produkt auf dem Markt mit dem entsprechenden Leistungsniveau erfüllt werden können.
- Sie müssen über eine technische Dokumentation verfügen, die das Produktdesign und die Anwendung, einschliesslich der Zweckbestimmung, verständlich macht, und die einen ausreichenden Nachweis liefert für die Konformität mit den geltenden allgemeinen Sicherheits- und Leistungsanforderungen (GSPR) in Anhang I der EU-MDR oder IVDR. (Gemäss der IVDR ist diese Anforderung nur für IVD der Klasse D verbindlich, die zuständigen Behörden können jedoch auch für niedrigere IVD-Klassen eine technische Dokumentation verlangen.)
- Sie müssen einer Überwachung nach dem Inverkehrbringen unterliegen und daraus resultierenden Korrekturmassnahmen.
- Nur für IVD-MDSW: Das Labor der Gesundheitseinrichtung muss die Norm EN ISO 15189 erfüllen oder relevante nationale Zulassungsvorschriften.
Die Verwendung von MDSW, die in Gesundheitseinrichtungen hergestellt werden, muss in einigen Ländern, darunter auch in der Schweiz, bei der zuständigen nationalen Behörde gemeldet werden.

MDSW, die ausschliesslich in einer Gesundheitseinrichtung hergestellt und verwendet werden, müssen nicht mit dem CE-Kennzeichen versehen werden.
Wie wird die korrekte Klassifizierung für MDSW gemäss EU-MDR oder IVDR festgelegt?
Bei der Klassifizierung von Medizinprodukten sind die MDCG-Leitlinien bzw. die Dokumente MDCG 2021-24 (Medizinprodukte) und MDCG 2020-16 (IVD) massgebend. Für Software reichen diese Dokumente jedoch nicht aus, auch wenn sie dort selbstverständlich abgedeckt ist und Beispiele genannt werden.
Für die Klassifizierung von MDSW ist der Leitfaden MDCG 2019-11 die nützlichste regulatorische Referenz für Hersteller, da es eine spezifische Auslegung (und anschauliche Beispiele) der Klassifizierungsregeln in Anhang VIII der EU-MDR und IVDR enthält.
Um Fehlklassifizierungen zu vermeiden, sollten die Durchführungsbestimmungen (und die zugehörigen Definitionen) sowie alle Klassifizierungsregeln im entsprechenden Anhang VIII überprüft werden. Dies ist besonders wichtig für MDSW, die als IVD eingestuft werden, da es in der IVDR keine softwarespezifischen Klassifizierungsregeln gibt und die gleichen gelten wie bei Hardware-IVDs.
Die erste Frage ist, ob die MDSW zusätzlich zu seinem eigenen medizinischen Zweck die Verwendung eines Hardware-Produkts oder -IVDs steuert oder beeinflusst. Wenn dies der Fall ist, fällt die MDSW unter die gleiche Klasse wie das Hardware-Produkt. Umgekehrt muss MDSW, die unabhängig von einem Hardware-Produkt ist, eigenständig klassifiziert werden.
Die nachstehenden Tabellen zeigen die Klassifizierungsregeln, die im Rahmen der EU-MDR und IVDR für MDSW zu beachten sind.
Für MDSW, die unter die EU-MDR fallen:
Klassifizierungsregeln für “aktive Produkte”, da Software als aktives Produkt gilt
|
Regel 9 |
Da Software nicht selbst physisch Energie abgeben oder austauschen kann, macht diese Regel nur für MDSW Sinn, die ein solches aktives Produkt steuert/beeinflusst. |
|
Regel 10 |
Da Software nicht direkt diagnostiziert oder überwacht, macht dies nur für MDSW Sinn, die ein solches aktives Produkt steuert/beeinflusst. |
|
Regel 11 |
Diese softwarespezifische Klassifizierungsregel existierte nicht unter der MDD. Sie wurde durch die EU-MDR eingeführt und ist die am häufigsten anwendbare Regel für Standalone-MDSW. |
|
Regel 12 |
Da Software nicht selbst Substanzen physisch verabreichen und/oder entfernen kann, macht diese Regel nur für MDSW Sinn, die ein solches aktives Produkt steuert/beeinflusst. |
|
Regel 13 |
Diese Regel gilt, wenn keine andere Regel anwendbar ist. Dies könnte für MDSW relevant sein, die ein aktives Produkt steuert/beeinflusst. |
Spezielle Klassifizierungsregeln
|
Regel 15 |
MDSW, die zur Empfängnisverhütung verwendet werden, fallen unter diese Regel und nicht unter die Regel 11. |
|
Regel 22 |
Diese Regel gilt für aktive therapeutische Produkte mit einer integrierten Diagnosefunktion, die massgeblich das Patientenmanagement durch das Produkt bestimmt, wie z.B. geschlossene Regelkreise oder automatische externe Defibrillatoren. Diese Vorschrift ist für MDSW relevant, die solche aktiven Produkte steuert/beeinflusst. |
Für MDSW, die unter die IVDR fallen (alle Klassifizierungsregeln können relevant sein):
Welche Arten von MDSW fallen unter die Klasse I der EU-MDR oder unter die Klasse A der IVDR?
Dies ist die am häufigsten gestellte und komplizierteste Frage im Zusammenhang mit MDSW.
Unter der früheren MDD wurde Standalone-Software gemäss der Klassifizierungsregel 12 in Anhang IX meist als Produkt der Klasse I eingestuft. Die neue Regel 11 in Anhang VIII der EU-MDR hat sich dramatisch auf die Klassifizierung ausgewirkt und führt meistens fast zwangsläufig zur Klasse IIa, was den Einbezug einer Benannten Stelle in den CE-Kennzeichnungprozess erforderlich macht. Darüber hinaus hat der Leitfaden MDCG 2019-11 in den Fällen, in denen der Hersteller gemäss der EU-MDR einen pragmatischen Ansatz für die Klassifizierung von MDSW hätte wählen können, eine starre Auslegung gebracht, die wenig Raum für MDSW der Klasse I lässt.
Die Klassifizierungsregel 11 der EU-MDR besteht aus drei Teilen:
- Für MDSW, die Informationen liefern, die die Grundlage für diagnostische oder therapeutische Entscheidungen bilden:
“Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:- death or an irreversible deterioration of a person’s state of health, in which case it is in class III; or
- a serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as class IIb.”
- Für MDSW, die Informationen in Bezug auf die Überwachung physiologischer Prozesse liefern, die letztlich auch zu diagnostischen oder therapeutischen Zwecken verwendet werden:
“Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.” - Für MDSW, die keinem der oben genannten Fälle entsprechen:
“All other software is classified as class I.”
Die MDCG 2019-11 weist darauf hin, dass Regel 11 eingeführt wurde, um das IMDRF-Rahmenwerk für die Kategorisierung von Software als Medizinprodukt, SaMD (IMDRF/SaMD WG/N12FINAL:2014), widerzuspiegeln. Dieses basiert auf der Kombination:
- der Bedeutung der von der Software bereitgestellten Informationen für die Entscheidung im Gesundheitswesen und
- der Gesundheitssituation oder dem Zustand des Patienten.
Die MDCG 2019-11 hat die IMDRF-Kategorisierung jedoch in einem konservativeren Licht neu interpretiert.
Die IMDRF wies Software, die das klinische Management bei nicht schwerwiegenden Erkrankungen steuert oder informiert, die niedrigste Risikokategorie zu:

die Fassung in Anhang III der MDCG 2019-11 schliesst MDSW der Klasse I aus:
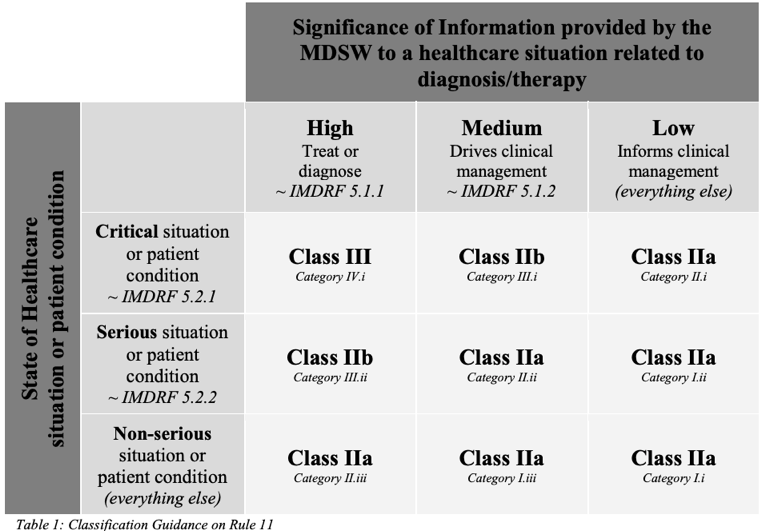
In der MDCG 2019-11 wird nur ein Beispiel für MDSW der Klasse I genannt: eine Mobile-App, welche die Empfängnis unterstützen soll, indem sie die Fruchtbarkeit durch Verfolgung und Vorhersage des Eisprungs auf der Grundlage eines statistischen Algorithmus berechnet.
Es könnten jedoch einige Schlupflöcher in Betracht gezogen werden:
- Eine MDSW, die von Patienten verwendet werden soll, um ihr Krankheitsrisiko auf der Grundlage der selbst eingegebenen Gesundheitsdaten abzuschätzen — z.B. ein Diabetes-Risikorechner. Eine solche MDSW könnte als nicht massgeblich oder informativ für das klinische Management angesehen werden, wenn der Output der Software beim Patienten verbleibt und nicht an eine medizinische Fachkraft weitergeleitet wird. Die vom Patienten selber eingegebenen Gesundheitsdaten können richtig oder falsch sein, und der Patient kann den Empfehlungen der Software folgen oder auch nicht.
- Der Begriff “therapeutische Zwecke” ist weder in der EU-MDR noch in der MDCG 2019-11 definiert. So könnte argumentiert werden, dass Zweckbestimmungen wie “Vorhersage” und “Prognose” (die in der EU-MDR neu in die Definition eines Medizinprodukts aufgenommen wurden) nicht therapeutischen Zwecken entsprechen. Dies würde heissen, dass MDSW mit als “other Software” gemäss Regel 11 gelten und somit in Klasse I fallen.
Was die IVDR anbelangt, so fällt kaum eine Software unter die Regel 5, die eine Einstufung in die die niedrigste Risikoklasse A ermöglicht, die einzige Regel, die keine Benannte Stelle im CE-Kennzeichnungsprozess erfordert. In der MDCG 2019-11 wird nur ein Beispiel für MDSW angeführt. Es handelt sich um eine Software, die ein Analysegerät zur Messung des C-reaktiven Proteins fernsteuert. Da das Analysegerät nach Regel 5 ein Produkt der Klasse A ist, ist auch die Software, die das Analysegerät betreibt, ein IVD der Klasse A.
Was ist für die CE-Kennzeichnung von MDSW gemäss der EU-MDR oder IVDR erforderlich?
Für MDSW gelten die gleichen grundlegenden Anforderungen wie für jedes andere Medizinprodukt oder In-vitro-Diagnostikum:
- Ein angemessenes Qualitätsmanagementsystem, d.h. mindestens gemäss EU-MDR oder IVDR Artikel 10. Produktklassen, die eine Zertifizierung durch eine Benannte Stelle erfordern, setzen ein Qualitätsmanagementsystem nach EN ISO 13485 voraus .
- Eine für die Einhaltung der Vorschriften verantwortliche Person (Person Responsible for Regulatory Compliance, PRRC), gemäss EU-MDR oder IVDR Artikel 15. Weitere Einzelheiten finden Sie in unserem Blogartikel Die Rolle des PRRC in der MDR, der auf die IVDR übertragen werden kann.
- Die technische Dokumentation gemäss EU-MDR oder IVDR Anhang II, die den Nachweis der Einhaltung der Allgemeinen Sicherheits- und Leistungsanforderungen (wie in Anhang I aufgeführt) erbringt, und gemäss Anhang III für die Überwachung nach dem Inverkehrbringen. Der Hauptnachweis für die Einhaltung der Anforderungen liegt in der Konformität mit der Norm EN 62304 über das MDSW-Lebenszyklusmanagement.
- Zuweisung einer eindeutigen Produktidentifikation (UDI). Weitere Einzelheiten finden Sie in unserem Blogartikel UDI für Medizinprodukte-Software unter der EU MDR.
- Zertifizierung durch eine Benannte Stelle für MDSW höher als Klasse I (EU-MDR) bzw. Klasse A (IVDR).
- Für Hersteller mit Sitz ausserhalb des EWR: Ein EU-Bevollmächtigter gemäss EU-MDR oder IVDR Artikel 11.
Weitere Einzelheiten zur CE-Kennzeichnung von Medizinprodukten und IVDs finden Sie in unseren separaten Blogartikeln:
- Wie man eine CE-Kennzeichnung für ein Medizinprodukt erhält
- CE-Kennzeichnung für In-vitro-Diagnostika nach der IVDR
Schliesslich sind die folgenden MDCG-Leitfäden von besonderer Bedeutung für die CE-Kennzeichnungsanforderungen von MDSW.
- MDCG 2020-1 über die klinische Bewertung oder Leistungsbewertung von MDSW
- MDCG 2019-16 zur Cybersicherheit
- MDCG 2018-5 zur UDI für MDSW
Für MDSW, die innerhalb einer Gesundheitseinrichtung entwickelt und verwendet werden, gelten vereinfachte Anforderungen. Siehe: Wird Software, die in einer Gesundheitseinrichtung entwickelt und verwendet wird, als MDSW betrachtet?
Ist die MDSW-Modularisierung ein geeigneter Ansatz?
Manchmal gelten nicht alle Funktionen/Teile einer bestimmten Software als MDSW. Ein Beispiel: Eine Software, die eine elektronische Patientenakte (nur Datenspeicherung und Datenkommunikation) mit einer Datenaggregation kombiniert, um eine Gesundheitswertung zu erstellen. Erreicht die Wertung bestimmte Schwellenwerte, sind klinische Massnahmen des Arztes erforderlich. Der Hersteller dieser MDSW kann sich dafür entscheiden, die Module, die nicht als MDSW gelten, vom CE-Kennzeichnungsprozess auszuschliessen.
Eine modulare Trennung ist möglich und wird in der MDCG 2019-11 in Betracht gezogen. In solchen Fällen ist der Hersteller dafür verantwortlich, die Grenzen und Schnittstellen der verschiedenen Module deutlich zu kennzeichnen. Dies kann ein verlockender Ansatz sein, um zu viel Arbeit zu vermeiden, insbesondere wenn die nichtmedizinischen Module von Dritten oder mit einem Ansatz entwickelt wurden, der für die Konformität mit der Norm EN 62304 nicht ausreicht.
In der Praxis erfordert der modulare Ansatz besondere Vorsicht bei Prozessen und Dokumentation, um Inkonsistenzen zu vermeiden. Einige Beispiele, wo er weniger geeignet ist, sind:
Kennzeichnung
Die Kennzeichnungsanforderungen von Medizinprodukten müssen auf der grafischen Benutzeroberfläche des MDSW implementiert werden. Üblicherweise geschieht dies im Menü “Über” oder auf dem Startbildschirm. Bei einem modularen Ansatz sind entweder separate “Über”-Menüs erforderlich, oder das einzige “Über”-Menü muss eindeutig angeben, welche Teile der Software MDSW sind und welche nicht.
Benutzerfreundlichkeit
Genau wie bei der Kennzeichnung hängt die Benutzerfreundlichkeit von der grafischen Benutzeroberfläche ab, und selbst wenn die Nicht-MDSW-Module deutlich von den MDSW-Modulen getrennt sind, werden bei den Tests zur Benutzerfreundlichkeit mit Sicherheit Informationen gesammelt, die für die MDSW-Gestaltung nicht relevant, aber für die allgemeine Benutzerfreundlichkeit wichtig sind und daher ebenfalls systematisch behandelt werden müssen.
Klinische Bewertung
Die Nicht-MDSW-Module können zu den klinischen Vorteilen der MDSW beitragen, z.B. wenn die direkte Kommunikation zwischen dem Patienten und dem Gesundheitsdienstleister zu einer schnellen therapeutischen Intervention führt. Der Ausschluss der Kommunikationsmodule aus der klinischen Bewertung führt zu einem verzerrten Bild der tatsächlichen klinischen Leistung der gesamten Software.
Umgang mit Beschwerden — PMS und Vigilanz
Ein ähnliches Problem ergibt sich bei der Notwendigkeit, den Grund für Beschwerden eindeutig den Modulen zuzuordnen. Je nach Software-Architektur ist dies gar nicht möglich, z.B. bei Schwachstellen in der Cybersicherheit, die sich auf die Datenübertragung auswirken, oder bei Fehlfunktionen der Datenspeicherung, die für die Diagnose oder therapeutische Intervention entscheidend sind.
Andere Aspekte der nichtmedizinischen Module können ebenso komplex sein. Es wird daher empfohlen, die Vor- und Nachteile gründlich zu analysieren, bevor man sich für einen Modularisierungsansatz entscheidet.
Kann ich mein MDSW als “Legacy-Produkt” auf den Markt bringen?
Für bestimmte Produkteklassen ist dies zwar möglich, aber die Beschränkung auf wesentliche Änderungen macht es schwierig.
Gemäss den Übergangsbestimmungen in EU-MDR Artikel 120 und IVDR Artikel 110 gelten MDSW, die mit der CE-Kennzeichnung versehen und im Rahmen der MDD oder IVDD auf dem EWR-Markt in Verkehr gebracht wurden, unter bestimmten Bedingungen als “Legacy-Produkte”. Zu diesen Bedingungen gehören:
Der “Legacy”-Status gilt nur für MDSW, die gemäss der EU-MDR oder IVDR die Beteiligung einer Benannten Stelle erfordert. Dies schliesst folgende MDSW aus: MDSW der Klasse I nach der MDD aus, die auch nach der EU-MDR die Klasse I bleiben würden (ein seltener Fall, wie bereits erwähnt), sowie selbstdeklarierte MDSW nach der IVDD, die nach der IVDR zur Klasse A würden (ebenfalls selten).
- Der “Legacy”-Status gilt nicht für MDSW der Klasse I gemäss MDD, die auch gemäss EU-MDR zur Klasse I gehört (ein seltener Fall, wie bereits erwähnt) oder für selbst deklarierte MDSW gemäss IVDD, die gemäss IVDR zur Klasse A gehören (ebenfalls selten).
- Es gibt keine wesentlichen Änderungen in Bezug auf das Produktdesign und Zweckbestimmung
Der Leitfaden MDCG 2020-3 enthält Einzelheiten zur Bedeutung von “wesentlichen Änderungen” im Zusammenhang mit Legacy-Produkten im Rahmen der EU-MDR. Es kann aber auch eine inspirierende Grundlage für die Bewertung von wesentlichen Änderungen im Rahmen der IVDR sein.
Im folgenden Blogartikel finden Sie mehr über MDCG 2020-3 und wesentliche Änderungen.
Abgesehen von Änderungen der Zweckbestimmung inklusive Einschränkungen/Erweiterungen der Zweckbestimmung, neuen Anwender-/Patientengruppen oder Änderungen der klinischen Anwendung, die immer als wesentlich angesehen werden, und Änderungen der Produktspezifikationen, führt die MDCG 2020-3 Tabelle C speziell Softwareänderungen auf, die als wesentlich betrachtet werden und zum Verlust des “Legacy”-Status führen:
- Neue oder wesentliche Änderung des Betriebssystems oder einer Komponente
- Neue oder geänderte Architektur oder Datenbankstruktur oder Änderung eines Algorithmus
- Ersetzen einer erforderlichen Benutzereingabe durch einen Algorithmus mit geschlossenem Regelkreis
- Neue diagnostische oder therapeutische Funktion oder neuer Kanal der Interoperabilität
- Neue Benutzerschnittstelle (UI) oder Datendarstellung (d.h. ein neues Format der Datendarstellung oder eine neue Dimension/Masseinheit)
Die obige Liste lässt nicht viel Raum für Softwareänderungen innerhalb der Grenzen der Legacy-Produkte. In der Tat sieht die MDCG 2020-3 nur wenige Fälle von Softwareänderungen vor, die nicht wesentlich wären, z.B.:
- Fehlerbehebungen, bei denen der behobene Fehler kein Sicherheitsrisiko birgt
- Sicherheitsaktualisierungen (z.B. Verbesserungen der Cybersicherheit, Berechnungen der Langlebigkeit)
- Erscheinungsbild der Benutzeroberfläche (z.B. neue Sprachen)
- Effizienzsteigerungen im Betrieb
- UI-Verbesserungen, die die Leistung nicht verändern
Angesichts der dynamischen Entwicklung von Software ist die Beibehaltung von MDSW als “Legacy” nur für gut etablierte klinische Anwendungen realistisch, bei denen Innovation keine Rolle spielt. Und schon gar nicht für solche, bei denen künstliche Intelligenz eingesetzt werden soll.
Weitere Einzelheiten zu Legacy-Produkten finden Sie in unserem Blogartikel “Legacy”- Medizinprodukte und -IVDs nach EU-Recht.
Wie Decomplix helfen kann
Decomplix bietet Ihnen eine fachkundige Beurteilung Ihrer Situation, der Qualifizierung und Klassifizierung Ihrer MDSW und einen vollständigen Fahrplan zur CE-Kennzeichnung gemäss EU-MDR oder IVDR. Hier können Sie mehr über unsere Dienstleistungen erfahren.
Weiter lesen
- Wie man eine CE-Kennzeichnung für ein Medizinprodukt erhält
- Ist mein Produkt in Europa ein Medizinprodukt? Wie Sie feststellen, ob Ihr Produkt die CE-Kennzeichnung für Medizinprodukte benötigt
- UDI für Medizinprodukte-Software unter der EU MDR
- “Legacy”- Medizinprodukte und -IVDs nach EU-Recht
- Welche Bedeutung hat die Zweckbestimmung bei Medizinprodukten?
- MDCG-Guidance zu wesentlichen Änderungen bei Medizinprodukten



