Medical device software (MDSW) under EU MDR and IVDR
Do you wonder whether your software is a medical device? If you are uncertain, read on. Do you develop software that is used for medical purposes? If so, and you are still unfamiliar with the EU MDR and IVDR, learn about CE-marking of medical device software (MDSW) here.
Replaces version of 02.03.2019
Key takeaways
- A “medical device” intended purpose is the determining factor in the qualification of software as MDSW, and the definition of medical device was extended in the EU MDR to introduce the purposes of prediction and prognosis. Yet, other lesser factors that make it into the equation might help avoid a MDSW qualification.
- Software classification has changed dramatically under the EU MDR and IVDR, with virtually no MDSW falling in low-risk classes and, thus, almost always requiring the involvement of a Notified Body in the CE marking process. Verify the classification rules to prevent misclassification and delays to market.
- “Legacy” status can apply to MDSW that was already on the market under the previous Directives. This comes with severe restrictions in changes that can be brought to the software. Make sure you understand the types of changes that would entail losing the “legacy” status.
Contents
What is Medical Device Software (MDSW)?
In summary, the following three conditions must be met for software to be qualified as MDSW:
- Having a medical purpose on its own,
- Performing an action on data beyond storage, archival, communication, simple search or lossless compression, and
- Being intended for the benefit of individual patients.
MDSW is software intended to be used (alone or in combination) for any of the purposes described in the definition of “medical device” in Article 2(1) of Regulation (EU) No. 2017/745 (EU MDR) or Regulation (EU) No. 2017/746 (IVDR).
The legal definition of “medical device” includes the following purposes:
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
- investigation, replacement or modification of the anatomy or of a physiological or pathological process or state,
- providing information by means of in vitro examination of specimens derived from the human body, including organ, blood and tissue donations,
- control or support of conception,
- cleaning, disinfection or sterilization of medical devices.
It is important to highlight that the EU MDR introduced a small but crucial extension to the definition of a medical device, in comparison with the former Directive 93/42/EEC (MDD), namely the purposes of prediction and prognosis of a disease. This means that software that might have not qualified as a medical device under the MDD, could now fall under the definition of medical device and, thus, need to comply with the requirements of the EU MDR or IVDR.
In brief, MDSW is software that has a medical device purpose on its own.
In other words, the intended purpose is a determining factor. You will see further below that it is not the only one, though. Still, not all healthcare software qualifies as a medical device. This is aligned with Recital (19) of the EU MDR: “software for general purposes, even when used in the healthcare setting, or software intended for life-style and well-being purposes is not considered a medical device”.
The type of action that the software performs on data is also relevant. According to MDCG 2019-11, software is not considered to be MDSW if such action is limited to:
- storage,
- archival,
- communication, understood to be “the flow of information from one point, known as the source, to another, the receiver”
- simple search, described as “the retrieval of records by matching record metadata against record search criteria or to the retrieval of information […] (e.g. library functions)”, or
- lossless compression, i.e. using a compression procedure that allows the exact reconstruction of the original data.
On the other hand, software intended to process, analyze, create or modify medical information may qualify as MDSW if these actions are governed by a medical intended purpose.
«In brief, MDSW is software that has a medical device purpose on its own.»
However, where those actions on data are not intended for the benefit of individual patients, for example, software that aggregates population data, provides generic diagnostic or treatment pathways, scientific literature (e.g. medical atlas), or software intended only for epidemiological studies or registries, such software is not considered MDSW either.
A software manufacturer can therefore avoid an MDSW qualification by acting on any of these three conditions.
It is also possible to separate the software functionality that would qualify as MDSW from the functionality that does not, with clearly distinct modules in the software architecture. For more details on a modular approach to MDSW, see Is MDSW modularization a suitable approach?
Now, the following types of software that do not have a medical device intended purpose by themselves, and are therefore not considered MDSW, are still subject to the requirements of the EU MDR or IVDR, as applicable:
- software that corresponds to the legal definition of “accessory” to either a medical device or an in-vitro diagnostic device (IVD),
- software that corresponds to products without intended medical, as listed in Annex XVI of the EU MDR,
- software that drives or influences the use of a (hardware) medical device or IVD, provided it does not have a separate medical intended purpose and does not create information on its own (in which case, the software module with separate medical intended purpose or generating information would qualify as MDSW).
In brief, MDSW is software that has a medical device purpose on its own.
When is MDSW an in-vitro diagnostic device (IVD)?
Software that qualifies as MDSW can fall under the EU MDR or IVDR. The applicable regulation is determined by the type of data processed. As such, MDSW is governed by the IVDR when:
- it provides information based on data obtained from IVDs only, or
- its intended purpose is substantially driven by IVD data sources.
For example, disease risk calculators where the inputs mainly come from blood and/or urine test results, would be MDSW that falls under the IVDR (“IVDR MDSW”). Conversely, disease risk calculators where the inputs come from electrocardiograms or imagery would be MDSW that falls under the EU MDR (“EU MDR MDSW”).
Now, software intended only to modify the representation of IVD results is not considered MDSW. MDCG 2019-11 provides the following examples of functionality that is considered to “modify the representation”:
- basic operations of arithmetic (e.g. mean, conversion of units),
- plotting of results in function of time,
- a comparison of the result to the limits of acceptance set by the user.
And, where the software is necessary and exclusively to render raw data from an IVD readable to the user, this would be MDSW driving or influencing the corresponding IVD.
How is MDSW different from Software as Medical Device (SaMD)?
SaMD is an international term, as defined by the IMDRF, that is carried over in the legislation of many geographies (including the UK), whereas MDSW is exclusively an EU term.
Specifically, IMDRF/SaMD WG/N10FINAL2013 defines SaMD as:
“Software intended to be used for one or more medical purposes that perform the purposes without being part of a hardware medical device. Software is a SaMD regardless of the fact that it needs hardware to execute. Regardless of whether it operates on general-purpose IT equipment, in “the cloud” or on the computing platform of a [hardware] of a medical device, as long as it is not “necessary for the [hardware] medical device to achieve its intended medical purpose.”
Whereas SaMD concerns only software that is independent and fully excludes any software “embedded” in a physical medical device, MDSW can be both independent or part of a hardware medical device, in the case where it has its own medical device purpose in addition to the purpose of driving/influencing the hardware medical device. A couple of examples of “embedded” software with own medical purpose are provided in MDCG 2019-11:
- Melanoma image analysis software intended to drive a near-infrared laser light scanner.
- MDSW intended to measure and transmit blood glucose levels, calculate insulin dose required.
Note also that the term “standalone” software that is widely used in the medtech industry is not a synonym of “independent” MDSW. A “standalone” MDSW can still drive or influence a hardware medical device.
Does software developed and used within a healthcare institution qualify as MDSW?
It may.
If such software has a medical device purpose on its own, is intended for the benefit of individual patients, and it processes data in a manner that is not only storage, archival, communication, lossless compression or simple search, it does qualify as MDSW.
«MDSW produced and used exclusively in a healthcare institution would not be CE-marked.»
Now, any device (including MDSW) that is produced within a healthcare facility and used solely within that facility is considered to have been put into service, per Article 5(4) in both the EU MDR and IVDR. Such devices are subject to the prerequisites specified in Art. 5(5) of the EU MDR or IVDR that are however far less extensive and cumbersome than the requirements applicable to devices placed on the market. MDSW produced and used exclusively in a healthcare institution would not be CE-marked still they must fulfill the following requirements:
- be designed/produced and used within an appropriate quality management system,
- have a justification issued by the health institution indicating that the target patient group’s specific needs cannot be met at the appropriate level of performance by an equivalent device on the market,
- have a technical documentation that allows understanding its design and deployment, including the intended purpose, and that provides sufficient evidence of compliance with the applicable General Safety and Performance Requirements (GSPR) in Annex I of the EU MDR or IVDR. Under the IVDR, this requirement is mandatory only for Class D IVDs but competent authorities might request a technical documentation also for lower IVD classes,
- be subject to post-market surveillance activities and, where necessary, to resulting corrective actions, and
- for IVD MDSW only, have the healthcare institution’s laboratory comply with EN ISO 15189 or any relevant national provisions on accreditation.
The use of MDSW produced in healthcare facilities requires notification to the national competent authority in some countries, including Switzerland.

MDSW produced and used exclusively in a healthcare institution would not be CE-marked.
How to establish the correct classification for MDSW under EU MDR or IVDR?
MDCG guidance documents on classification of medical devices (MDCG 2021-24) and of IVDs (MDCG 2020-16) are the ones to be followed when classifying devices. Yet, even if software is obviously covered and examples are provided, these documents alone do not suffice.
Guidance document MDCG 2019-11 is the most useful regulatory reference for manufacturers in the classification of MDSW, as it provides specific interpretation (and illustrative examples) of the classification rules in Annex VIII of the EU MDR and IVDR.
When classifying a medical device or IVD, the implementing rules (and related definitions) as well as all classification rules in the corresponding Annex VIII should be reviewed to avoid misclassification. This is particularly important for MDSW qualifying as IVD, since there are no software-specific classification rules in the IVDR and the same classification rules as for hardware IVDs must be followed.
The first question is whether MDSW drives or influences the use of a (hardware) device or IVD, in addition to having its own intended medical purpose. If so, such MDSW falls under the same class as the (hardware) device. Conversely, MDSW that is independent from a (hardware) device must be classified in its own right.
The two tables below indicate the classification rules that need attention under the EU MDR and IVDR for MDSW.
For MDSW falling under the EU MDR:
Classification rules for “active devices”, since software is considered to be an active device
|
Rule 9 |
Because software cannot physically administer or exchange energy (incl. those that monitor or control other active devices), this rule could only apply to MDSW driving/influencing active devices that administer/exchange energy. |
|
Rule 10 |
Because software cannot directly diagnose or monitor, this could only apply to MDSW driving/influencing such an active device. |
|
Rule 11 |
This is a new, software-specific classification rule, introduced by the EU MDR, i.e. it did not exist under the MDD, and it is the most frequently applicable rule for independent MDSW. |
|
Rule 12 |
Because software cannot physically administer and/or remove substances, this rule could only apply to MDSW driving/influencing such an active device. |
|
Rule 13 |
If no other rule applies, this rule would apply. This could be relevant for MDSW driving/influencing an active device. |
Special classification rules
|
Rule 15 |
MDSW used for contraception would fall under this rule instead of Rule 11. |
|
Rule 22 |
This rule covers active therapeutic devices with an integrated or incorporated diagnostic function which significantly determines the patient management by the device, such as closed loop systems or automated external defibrillators. |
For MDSW falling under the IVDR (all classification rules might be relevant):
|
Rule 1 – First indent |
Because software cannot physically detect transmissible agents, this classification sub-rule could only apply to MDSW driving/influencing such IVDs. |
|
Rule 1 – Second indent |
Because software cannot physically detect transmissible agents, this classification sub-rule could only apply to MDSW driving/influencing such IVDs. |
|
Rule 1 – Third indent |
Because software cannot physically determine an infectious load, this classification sub-rule could only apply to MDSW driving/influencing such IVDs. |
|
Rule 2 |
Because software cannot physically determine blood groups, this rule could only apply to MDSW driving/influencing such IVDs. |
|
Rule 3 |
This rule encompasses 13 sub-rules that involve the physical analysis of specimens collected from the human body. |
|
Rule 4 |
Because software cannot physically test body samples, this rule could only apply to MDSW driving/influencing such IVDs. |
|
Rule 5 (a) |
This classification sub-rule applies to general laboratory products like pipettes, stain powders, glass microscope slides, centrifuges, pipette tips or instrument liquid collection containers, buffers which usually do not fall under the definition of an IVD medical device. |
|
Rule 5 (b) |
Because software cannot physically run any in-vitro analysis, this classification sub-rule could only apply to MDSW driving/influencing such IVDs. |
|
Rule 5 (c) |
Because software cannot physically be a receptacle, this rule is unlikely to apply. |
|
Rule 6 |
This rule would be the most frequently applicable for independent MDSW, particularly software that uses IVD data for further interpretation or analysis. |
|
Rule 7 |
Because software cannot physically be a control material, this rule could only apply to MDSW driving/influencing such IVDs. |
What types of MDSW would classify as Class I under EU MDR or as Class A under IVDR?
This is the most frequently asked and most convoluted question relative to MDSW.
Under the former MDD, “standalone” software was mostly classified as a class I device, per classification Rule 12 in Annex IX, since no specific classification rules applied to software. The new Rule 11 in EU MDR Annex VIII has dramatically impacted classification and almost inevitably leads to, at a minimum, class IIa, thus requiring the involvement of a Notified Body in the conformity assessment for CE-marking. Furthermore, wherever the manufacturer could have taken a pragmatic approach to MDSW classification under the EU MDR, guidance document MDCG 2019-11 brought a rigid interpretation that leaves little room for Class I MDSW.
EU MDR’s classification Rule 11 contains three parts:
- For MDSW that provides information which is the basis for diagnostic or therapeutic decisions:
“Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause:- death or an irreversible deterioration of a person’s state of health, in which case it is in class III; or
- a serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as class IIb.”
- For MDSW that provides information relative to the monitoring of physiological processes, which ultimately would also be used for diagnostic or therapeutic purposes:
“Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb.” - For MDSW that does not correspond to any of the above cases:
“All other software is classified as class I.”
MDCG 2019-11 indicates that Rule 11 was introduced to mirror the IMDRF framework for the categorization of software as a medical device, SaMD (IMDRF/SaMD WG/N12FINAL:2014), which is based on the combination of:
- the significance of the information provided by the software to the healthcare decision, and
- the healthcare situation or patient condition.
However, MDCG 2019-11 has reinterpreted the IMDRF’s categorization under a more conservative light.
Whereas IMDRF assigned the lowest-risk category to software that drives or informs clinical management in case of non-serious conditions:

the version in Annex III of the MDCG 2019-11 excludes Class I MDSW:
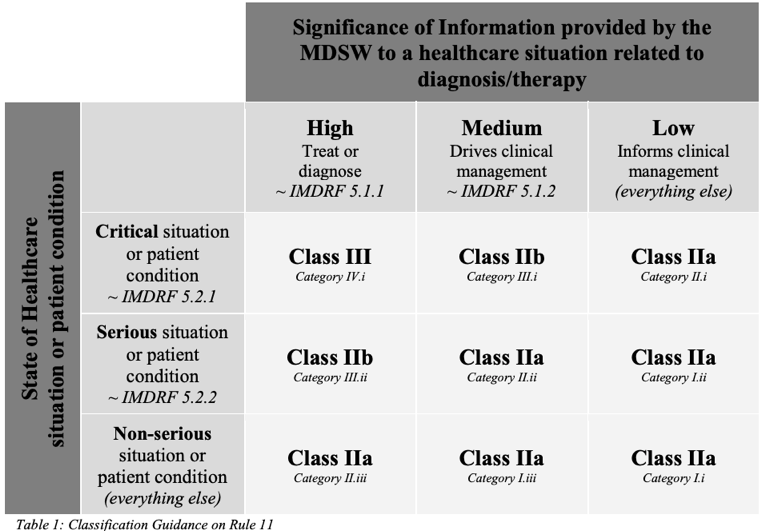
MDCG 2019-11 cites only one example of Class I MDSW: a mobile app intended to support conception by calculating the user’s fertility status by tracking and predicting ovulation, based on a validated statistical algorithm.
Yet, a couple of loopholes could be envisaged:
- MDSW that is intended for use by patients to estimate their own risk of suffering a given condition based on health data entered by the patient, e.g. a diabetes risk calculator, could be considered not to drive or inform clinical management if the software output remains with the patient and is not transferred to a healthcare professional. The patient might or might not follow the recommendations that the software yields after processing the clinical data provided by the patient as input, which in turn may or may not be correct.
- The term “therapeutic purposes” is not defined in the EU MDR or in MDCG 2019-11 and, thus, it could be argued that intended purposes like prediction and prognosis (newly introduced in the definition of a medical device under the EU MDR) do not correspond to “therapeutic purposes”. This could leave an option for MDSW with such purposes to correspond to “other software” under Rule 11, thus falling into Class I.
As to the IVDR, hardly any software falls under Rule 5, which is the only rule yielding a Class A classification, i.e. the lowest risk class that does not require Notified Body involvement in the CE-marking process. MDCG 2019-11 only cites one example for MDSW driving a C-reactive protein measuring analyzer from a remote location, since the analyzer would be a Class A device under Rule 5, the software operating the analyzer would also be a Class A IVD.
What is needed to CE-mark MDSW under the EU MDR or IVDR?
The same basic requirements as for any other medical device under the EU MDR or for any in-vitro diagnostic device under the IVDR apply to MDSW, in brief:
- An appropriate quality management system, per EU MDR or IVDR Article 10 at a minimum. For device classes requiring Notified Body certification, an EN ISO 13485 compliant quality management system would be a prerequisite.
- A Person Responsible for Regulatory Compliance (PRRC), per EU MDR or IVDR Article 15. For more details, see our blog article The role of the PRRC under the MDR, which can be extrapolated to the IVDR.
- The technical documentation according to EU MDR or IVDR Annex II, providing evidence of compliance with the General Safety and Performance Requirements (GSPR, as listed in Annex I), and Annex III for post-market surveillance. The core evidence of GSPR compliance would have to be based on conformity with standard EN 62304 on medical device software lifecycle management.
- Unique Device Identification (UDI) assignment, as applicable to MDSW. For more details, see our blog article UDI for Medical Device Software (MDSW) under EU MDR.
- Certification by a Notified Body, for MDSW above Class I under the EU MDR or Class A under the IVDR.
- For manufacturers based outside of EEA, an EU Authorized Representative, per EU MDR or IVDR Article 11.
For more details on CE-marking medical devices and IVDs, see our separate blog articles:
- How to obtain a CE mark for a medical device
- CE marking for in vitro diagnostic devices under the IVDR
Last, the below MDCG guidance documents are of particular importance for CE-marking requirements of MDSW.
- MDCG 2020-1 on the clinical evaluation or performance evaluation of MDSW
- MDCG 2019-16 on cybersecurity
- MDCG 2018-5 on UDI for MDSW
For MDSW developed and used within a healthcare institution, there are simplified requirements. See: Does software developed and used within a healthcare institution qualify as MDSW?
Is MDSW modularization a suitable approach?
Sometimes, not all functions/modules of a given software qualify as MDSW. For example, a software that combines electronic health record functionality (data storage and data communication functions only) with data aggregation to establish a health score where certain thresholds require clinical action by the physician. MDSW manufacturers may then choose to exclude the modules that do not qualify as MDSW from the CE-marking process.
A modular segregation is possible and contemplated in MDCG 2019-11. In such cases, the manufacturer is responsible for distinctly identifying the boundaries and interfaces of the different modules. It might seem a compelling approach to avoid too much work, particularly when the non-medical modules have been developed by third parties or with an approach that is not sufficiently rigorous for EN 62304 conformity.
In practice, the modular approach requires extra caution across processes and documentation to prevent inconsistencies. Some examples that make the modular approach less convenient are:
Labelling
Medical device labelling requirements have to be implemented on the MDSW’s graphic user interface, usually the ”about” menu or the start-up screen. In a modular approach, either separate “about” menus are needed or the single ”about” menu must clearly identify which parts of the software are MDSW and which parts are not.
Usability
Just like for labelling, software usability resides on the graphic user interface and, even when the non-MSDW modules are distinctly separated from the MDSW modules, usability testing will certainly collect inputs that might not be relevant for MDSW design but important for the overall user satisfaction and, thus, need to be addressed in systematic manner, too.
Clinical evaluation
The non-MDSW modules may have a contributory effect to the clinical benefits of the MDSW, e.g. where direct communication between the patient and the healthcare provider leads to a rapid therapeutic intervention. Excluding the communication modules from the clinical evaluation results in a biased picture of the actual clinical performance of the entire software.
Complaint handling, PMS and Vigilance
A same problem arises on the need to clearly differentiate the root cause of complaints as being related to the MDSW modules or to the non-MDSW modules. Depending on the software architecture, this might not even be feasible, e.g. cybersecurity vulnerabilities affecting data transfer functionality or data storage malfunction that are critical for diagnosis or therapeutic intervention.
Other aspects of non-medical modules can be equally complex. It is therefore recommended to thoroughly analyze the pros and cons before taking a modularization approach.

In practice, the modular approach requires extra caution across processes and documentation to prevent inconsistencies.
Can I keep my MDSW as a “legacy device”?
Although it is possible for certain device classes, the limitation on significant changes makes it difficult.
Pursuant to the transitional provisions in EU MDR Art. 120 and IVDR Art. 110, MDSW that was duly CE-marked and placed on the EEA market under the MDD or IVDD qualifies as “legacy” device, under certain conditions. Amongst these:
- The “legacy” status may apply only to MDSW that will require Notified Body involvement under the EU MDR or IVDR. This excludes Class I MDSW under the MDD that would remain also Class I under the EU MDR (a rare case, as discussed earlier) as well as self-declared MDSW under the IVDD that would become Class A under the IVDR (also rare).
- There are no significant changes in the design and intended purpose.
Guidance document MDCG 2020-3 provides details on the meaning of “significant changes” in the context of “legacy” devices under the EU MDR, but it can also be an inspiring basis for evaluating significant changes under the IVDR (read more about MDCG 2020-3 and significant changes).
Other than changes in intended purpose (incl. limitations/extensions of intended purpose, new user/patient population or changes in clinical use), which are always considered to be significant, and changes to the device specifications, MDCG 2020-3 Chart C specifically details software changes that would be viewed as significant and would entail losing the “legacy” status:
- New or major change of operating system or any component
- New or modified architecture or database structure or change of an algorithm
- Replacing a required user input by a closed-loop algorithm
- New diagnostic or therapeutic feature or new channel of interoperability
- New user interface (UI) or presentation of data (which refers to new data representation format or new dimension or measuring unit)
The above list does not leave much room for software changes within the “legacy” device boundaries. Indeed, MDCG 2020-3 sees only few cases of software changes that would not be significant, for example:
- Bug fixes, where the corrected error does not pose a safety risk
- Security updates (e.g. cybersecurity enhancements, longevity calculations)
- UI appearance (which could include new languages)
- Operating efficiencies
- UI enhancements that do not change performance
Given the dynamic evolution of software, keeping MDSW as “legacy” is only realistic for well-established clinical applications where innovation is not a driver. Definitely not for those where artificial intelligence is an emerging option.
For more details on “legacy” devices, please see our blog article “Legacy” medical devices and IVDs under EU legislation.
How Decomplix can help
Decomplix provides expert assessment of your situation, the qualification and classification of your MDSW, and a complete roadmap to obtaining a CE mark for your MDSW under the EU MDR or IVDR. You can learn more about our services here.
Further reading
- How to obtain a CE mark for a medical device
- Is my product a medical device in Europe? How to determine if your product requires medical device CE marking
- UDI for Medical Device Software (MDSW) under EU MDR
- “Legacy” medical devices and IVDs under EU legislation
- The intended purpose – or, what does your medical device do?
- Guidance on medical device significant changes



