
Ist mein Produkt in Europa ein Medizinprodukt? Wie Sie feststellen, ob Ihr Produkt die CE-Kennzeichnung für Medizinprodukte benötigt
Wenn Sie wissen wollen, ob Ihr Produkt in der EU als Medizinprodukt oder In-vitro-Diagnostikum (IVD) gilt, und ob es somit eine CE-Kennzeichnung als Medizinprodukt oder IVD benötigt, lesen Sie weiter. Die Antwort auf eine scheinbar einfache Frage kann komplex sein.
Ersetzt die Version vom 12.02.2019
Wichtige Erkenntnisse:
- Auch Zubehör, Komponenten und Produkte, die nur ästhetischen Zwecken dienen, können als Medizinprodukte gelten.
- Um eine korrekte Qualifizierung zu erreichen, Berücksichtigen Sie bei der Beurteilung der regulatorischen Kategorie Ihres Produkts alle rechtlichen Definitionen und Ausschlüsse vom Anwendungsbereich in der EU-MDR und IVDR. Verwenden Sie das Flussdiagramm in diesem Artikel zur Qualifizierungsentscheidung.
- Bei Produkten, die ein Grenzfall sind, s.g. Borderline-Produkten, sollten Sie sich nicht auf Ihre eigene Interpretation verlassen. Ziehen Sie die offiziellen Leitlinien und gegebenenfalls sogar die Urteile des Europäischen Gerichtshofs zu Rate.
Inhalt:
Woran erkennt man, ob es sich um ein Medizinprodukt nach EU-MDR handelt? Der Ausgangspunkt
Die Bestimmung des für Ihr Produkt geltenden Rechtsrahmens kann eine Herausforderung darstellen. Einige Produkte lassen sich nicht ohne Weiteres einordnen, weil sie Funktionen kombinieren, die verschiedenen Regelwerken entsprechen, oder weil sie aufgrund ihrer Beschaffenheit ein Grenzfall sind und unter das eine oder andere Regelwerk fallen können. In solchen Situationen beruht die endgültige Entscheidung auf einer Einzelfallprüfung.
Im ersten Schritt müssen Sie vom Zweck ausgehen, den Sie Ihrem Produkt zuweisen, und diesen hinreichend genau beschreiben. So wird klar, ob es sich bei der Verwendung Ihres Produkts um eine medizinische Massnahme handelt oder nicht. Z.B. kann eine Pinzette den Zweck haben, Splitter aus einer Wunde zu entfernen (medizinischer Zweck), Augenbrauen zu zupfen (kosmetischer Zweck) oder Fischgräten zu entfernen (allgemeines Verbraucherprodukt).
Als Faustregel gilt: Wenn Sie für Ihr Produkt einen medizinischen Zweck vorsehen, stehen die Chancen gut, dass es sich um ein Medizinprodukt bzw. IVD handelt. Ein genauer Blick auf die rechtsverbindlichen Definitionen ist jedoch unerlässlich.
Inwiefern ist der rechtliche Rahmen für die Qualifizierung von Medizinprodukten hilfreich?
Der derzeitige Rechtsrahmen im Europäischen Wirtschaftsraum (EWR), der Türkei und der Schweiz basiert auf der Verordnung (EU) 2017/745 der Europäischen Union über Medizinprodukte (EU-MDR) und der Verordnung (EU) 2017/746 über In-vitro-Diagnostika (IVDR). Beide Verordnungen gelten in allen EU-Mitgliedstaaten in ihrer jetzigen Form und werden in anderen EWR-Ländern, der Türkei und der Schweiz in unterschiedlichem Masse durch nationale Gesetzgebung übernommen. Zur Vereinfachung beziehen wir uns in diesem Artikel allgemein auf die EU.
Die EU-MDR enthält rechtsverbindliche Definitionen von Medizinprodukten und Zubehör sowie Ausnahmen vom Anwendungsbereich der Verordnung zur Abgrenzung von Medizinprodukten zu anderen eng verwandten Produkten (z.B. Arzneimittel, Produkte für neuartige Therapien, biologische Komponenten, Kosmetika oder Lebensmittel). Darüber hinaus enthält die EU-MDR in Anhang XVI eine Liste von Produkten ohne medizinische Zweckbestimmung, wofür die Verordnung ebenfalls gilt.
Die IVDR enthält die spezifischen Definitionen für IVD, sowie für IVD-Zubehör, und die entsprechenden Ausnahmen vom Anwendungsbereich der Verordnung zur Abgrenzung von IVDs zu Medizinprodukten, aber auch zu allgemeinen Laborgeräten oder reinen Forschungsgeräten.
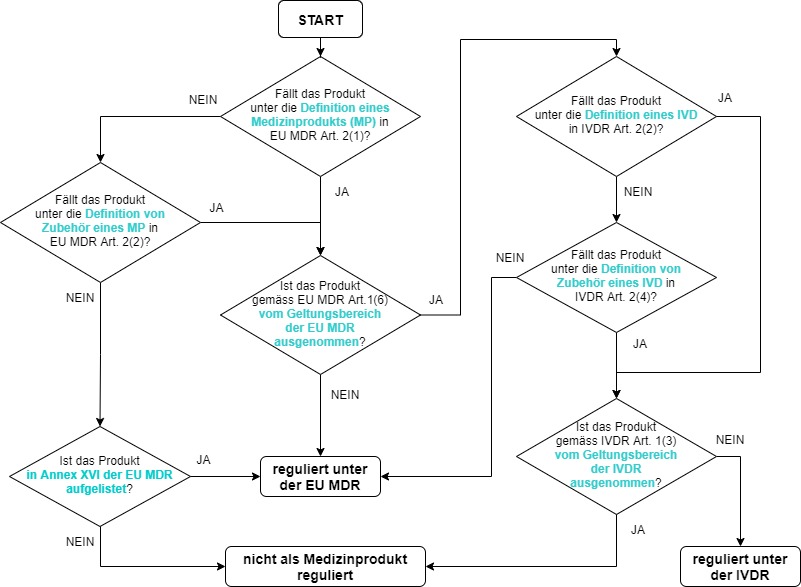
Nimmt man beide Verordnungen zusammen, so ergeben sich für die Qualifizierung von Medizinprodukten oder IVD folgende Fragen:
- Fällt das Produkt unter die Definition eines Medizinprodukts?
- Fällt das Produkt unter die Definition von Zubehör für ein Medizinprodukt?
- Ist das Produkt in Anhang XVI der EU-MDR aufgeführt?
- Ist das Produkt vom Geltungsbereich der EU-MDR ausgenommen?
- Fällt das Produkt unter die Definition eines IVD?
- Fällt das Produkt unter die Definition von Zubehör für ein IVD?
- Ist das Produkt vom Anwendungsbereich der IVDR ausgenommen (und handelt es sich nicht um ein Produkt zur invasiven Probenahme)?
Diese Fragen können mit dem nachstehenden Flussdiagramm für Qualifikationsentscheidungen durchlaufen werden.
Die erste rechtliche Definition, die Sie demnach lesen müssen, ist diejenige des Medizinprodukts. EU-MDR Art 2, Abs 1:
„Medizinprodukt“ bezeichnet ein Instrument, einen Apparat, ein Gerät, eine Software, ein Implantat, ein Reagenz, ein Material oder einen anderen Gegenstand, das dem Hersteller zufolge für Menschen bestimmt ist und allein oder in Kombination einen oder mehrere der folgenden spezifischen medizinischen Zwecke erfüllen soll:
- Diagnose, Verhütung, Überwachung, Vorhersage, Prognose, Behandlung oder Linderung von Krankheiten,
- Diagnose, Überwachung, Behandlung, Linderung von oder Kompensierung von Verletzungen oder Behinderungen,
- Untersuchung, Ersatz oder Veränderung der Anatomie oder eines physiologischen oder pathologischen Vorgangs oder Zustands,
- Gewinnung von Informationen durch die In-vitro-Untersuchung von aus dem menschlichen Körper — auch aus Organ-, Blut- und Gewebespenden — stammenden Proben
und dessen bestimmungsgemässe Hauptwirkung im oder am menschlichen Körper weder durch pharmakologische oder immunologische Mittel noch metabolisch erreicht wird, dessen Wirkungsweise aber durch solche Mittel unterstützt werden kann.
Die folgenden Produkte gelten ebenfalls als Medizinprodukte:
- Produkte zur Empfängnisverhütung oder -förderung,
- Produkte, die speziell für die Reinigung, Desinfektion oder Sterilisation der in Art. 1 Abs. 4 genannten Produkte und der in Absatz 1 dieses Spiegelstrichs genannten Produkte bestimmt sind.
Die Definition stützt sich also auf die vom Hersteller vorgesehene Zweckbestimmung. Sie ist das entscheidende Element der Qualifizierung. Eine Nuancierung wird durch die Hauptwirkungsweise des Produkts erreicht, um eine Abgrenzung zu Arzneimitteln zu ermöglichen, die die nächstliegenden Grenzprodukte darstellen.
Wenn Ihr Produkt keinen der oben genannten medizinischen Zwecke erfüllt, kann es dennoch als Medizinprodukt im Sinne der EU-MDR angesehen werden, da auch Zubehör und Produkte nach Anhang XVI berücksichtigt werden müssen.
“Zubehör” ist ein Begriff, der sowohl in der EU-MDR als auch in der IVDR rechtlich definiert ist.
– Zubehör für Medizinprodukte bedeutet gemäss EU MDR Art. 2 Abs 2: „Zubehör eines Medizinprodukts“ bezeichnet einen Gegenstand, der zwar an sich kein Medizinprodukt ist, aber vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren bestimmten Medizinprodukten verwendet zu werden, und der speziell dessen/deren Verwendung gemäss seiner/ihrer Zweckbestimmung(en) ermöglicht oder mit dem die medizinische Funktion des Medizinprodukts bzw. der Medizinprodukte im Hinblick auf dessen/deren Zweckbestimmung(en) gezielt und unmittelbar unterstützt werden soll;
– Ähnliches gilt für Zubehör für IVD gemäss Art 2 Abs 4 der IVDR: „Zubehör eines In-vitro-Diagnostikums“ bezeichnet einen Gegenstand, der zwar an sich kein In-vitro-Diagnostikum ist, aber vom Hersteller dazu bestimmt ist, zusammen mit einem oder mehreren bestimmten In-vitro-Diagnostika verwendet zu werden, und der speziell dessen/ deren Verwendung gemäss seiner/ihrer Zweckbestimmung(en) ermöglicht oder mit dem die medizinische Funktion des In-vitro-Diagnostikums/der In-vitro-Diagnostika im Hinblick auf dessen/deren Zweckbestimmung(en) gezielt und unmittelbar unterstützt werden soll;
Um festzustellen, ob Ihr Produkt unter die Definition von “Zubehör” fällt oder ob es sich um eine Art Gebrauchsgegenstand handelt, der nicht zusammen mit einem Medizinprodukt oder IVD verwendet werden soll, müssen Sie genau hinschauen. Auch hier kommt es auf die Zweckbestimmung des Produkts an.
Ebenfalls berücksichtigen müssen Sie Produktteile oder Komponenten. Diese werden von den Herstellern in der Regel als Ersatzteile oder optionale Teile verstanden, die getrennt vom Produkt geliefert werden, mit dem sie verwendet werden sollen. Sowohl nach der EU-MDR als auch nach der IVDR gelten Teile und Komponenten, die die Leistungs- oder Sicherheitsmerkmale oder die Zweckbestimmung eines Medizinprodukts erheblich verändern, ebenfalls als Medizinprodukte.
Darüber hinaus wurde eine neue Kategorie von Produkten in den Anhang XVI der EU-MDR aufgenommen. Dabei handelt es sich hauptsächlich um Produkte und Gegenstände für ästhetische Zwecke, die bisher nicht geregelt waren und nun den Anforderungen an Medizinprodukte entsprechen müssen. Dazu gehören:
- Kontaktlinsen oder andere Gegenstände, die dazu bestimmt sind, in das Auge eingeführt bzw. auf das Auge aufgetragen zu werden.
- Chirurgisch invasive Produkte zur Veränderung der Anatomie oder zur Fixierung von Körperteilen (ausgenommen Tätowiermittel und Piercings).
- Stoffe, Stoffkombinationen oder Gegenstände, die zum Auffüllen des Gesichts oder anderer Haut- oder Schleimhäute bestimmt sind (ausgenommen Tätowiermittel).
- Produkte zur Reduzierung, Entfernung oder Zerstörung von Fettgewebe (z. B. Produkte zur Fettabsaugung, Lipolyse oder Lipoplastik).
- Produkte mit hochintensiver elektromagnetischer Strahlung (z.B. Infrarot, sichtbares Licht und Ultraviolett), die für die Hauterneuerung, die Entfernung von Tätowierungen oder Haaren oder andere Hautbehandlungen bestimmt sind.
- Produkte zur Hirnstimulation, die elektrische Ströme oder magnetische oder elektromagnetische Felder anwenden, die den Schädel durchdringen, um die neuronale Aktivität des Gehirns zu verändern.
Wenn Ihr Produkt eine In-vitro-Anwendung hat, d.h. seine Zweckbestimmung dreht sich um die Untersuchung von Körperproben (z.B. Blut, Speichel, Urin), sollten Sie die IVD-Definition in Art 2 Abs 2 der IVDR konsultieren, um sicherzustellen, dass Sie die richtige Verordnung auswählen. Beachten Sie dies:
- Ein Produkt kann nicht allein durch die Angabe “zur Verwendung in der In-vitro-Diagnostik” in den Geltungsbereich der IVDR fallen; es muss einen medizinischem Zweck haben.
- Gegenstände, die zur invasiven Entnahme von Körperproben oder zur Entnahme von Proben von der Körperoberfläche bestimmt sind (z.B. Lanzetten, Tupfer), gelten als Medizinprodukte und fallen unter die EU-MDR.
- Allgemeine Laborgeräte und reine Forschungsprodukte gelten nicht als IVD und werden daher als allgemeine Verbraucherprodukte betrachtet.
Auch Zubehör, Komponenten und Produkte, die nur ästhetischen Zwecken dienen, gelten möglicherweise als Medizinprodukte und müssen mit der CE-Kennzeichnung versehen werden.
Fällt Ihr Produkt unter die Definition eines Medizinprodukts in Art 2 Abs 1 der EU-MDR und/oder unter die Definition eines IVD in Art 2 Abs 2 der IVDR, kann es trotzdem von deren Anwendungsbereich ausgenommen sein und unter eine andere Regelung fallen. Zum Beispiel, wenn das Produkt die entsprechende gesetzliche Definition erfüllt:
- In der Praxis sind Kombinationsprodukte aus Arzneimitteln und Medizinprodukten nicht immer leicht einzuordnen. Oft werden sie durch die Richtlinie 2001/83/EG für Arzneimittel geregelt.
- Produkte für neuartige Therapien werden durch die Verordnung (EG) Nr. 1394/2007 geregelt.
- Transplantate, Gewebe/Zellen menschlichen Ursprungs und ihre Derivate werden durch die Richtlinie 2004/23/EG geregelt. Medizinprodukte (oder Zubehör oder Produkte nach Anhang XVI), die unter Verwendung von Geweben/Zellen menschlichen Ursprungs hergestellt werden, fallen jedoch weiterhin in den Geltungsbereich der EU-MDR.
- Kosmetika werden durch die Verordnung (EG) Nr. 1223/2009 geregelt.
- Lebensmittel werden durch die Verordnung (EG) Nr. 178/2002 geregelt.
Wie Sie sehen, ist die Qualifizierung nur bei sehr offensichtlichen Medizinprodukten einfach. Ist dies bei Ihrem Produkt nicht der Fall, so empfehlen wir Ihnen, weiterzulesen.
Borderline-Produkte: Wie lässt sich die Unsicherheit bewältigen?
Trotz der gesetzlichen Definitionen und der Ausschlüsse vom Anwendungsbereich gibt es immer noch Fälle, die zwischen den Regulierungen liegen. Zum Beispiel: Ist ein Desinfektionsmittel ein Arzneimittel, ein Medizinprodukt oder ein Biozid? Nun, das hängt davon ab, was es desinfizieren soll und wie die desinfizierende Wirkung erzielt wird.
Es gibt eine Reihe von Instrumenten, um die Unsicherheit bei der Einstufung dieser Produkte zu bewältigen. Ein häufiger Fehler ist, sich auf die eigene Interpretation einer Grenzsituation zu verlassen, ohne weiter nach einer offiziellen regulatorischen Position zu suchen.
Eine der umfassendsten Leitlinien im Rahmen der früheren EU-Gesetzgebung war das Manual on Borderline and Classification der Europäischen Kommission, das zuletzt im Mai 2019 aktualisiert wurde. Darin wurden Beispiele für Borderline-Produkte und die von den EU-Regulierungsbehörden vereinbarten und geteilten Schlussfolgerungen zusammengestellt. Ein entsprechendes Manual on Borderline and Classification under the EU MDR and IVDR wurde im September 2022 gemeinsam von der Borderline & Classification Working Group und der Medical Device Coordination Group (MDCG) veröffentlicht. Diese erste Version enthält nur einige wenige Fälle und wird weiter ergänzt werden, sobald weitere Entscheidungen getroffen worden sind. Wie bei allen offiziellen Leitlinien sind die in diesem Handbuch dargelegten Standpunkte nicht rechtsverbindlich. Denn nur der Europäische Gerichtshof (EuGH) ist berechtigt, eine verbindliche Auslegung von Fragen der Qualifikation von Medizinprodukten, wie auch von anderen Aspekten der EU-Gesetzgebung, vorzunehmen.
Das neue Manual on Borderline and Classification ist in zwei Abschnitte gegliedert, die jeweils die EU-MDR und die IVDR abdecken, mit Unterabschnitten zu Fragen der Qualifikation und der Klassifizierung.
Der Teilbereich EU-MDR-Qualifikation umfasst 9 Kapitel, die in verschiedene Abschnitte unterteilt sind und aufzeigen, wo Schwierigkeiten liegen:
- Kapitel 1.1.1: Abgrenzung zwischen Medizinprodukten und IVDs. In diesem Kapitel werden die Fälle behandelt, in denen die Schlussfolgerung lautet, dass das Produkt als Medizinprodukt eingestuft werden sollte. Produkte, die als IVD eingestuft werden sollten, werden in Kapitel 2.1.1 behandelt.
- Kapitel 1.1.2: Abgrenzung zwischen Medizinprodukten und Arzneimitteln, einschliesslich Arzneimitteln für neuartige Therapien (ATMP). Es wurde auch ein separater MDCG-Leitfaden veröffentlicht (siehe unten).
- Kapitel 1.1.3: Abgrenzung zwischen Medizinprodukten und Bioziden.
- Kapitel 1.1.4: Abgrenzung zwischen Medizinprodukten und Stoffen menschlichen Ursprungs.
- Kapitel 1.1.5: Abgrenzung zwischen Medizinprodukten und kosmetischen Produkten.
- Kapitel 1.1.6: Abgrenzung zwischen Medizinprodukten und Lebensmitteln.
- Kapitel 1.1.7: Abgrenzung zwischen Medizinprodukten und persönlicher Schutzausrüstung.
- Kapitel 1.1.8: Abgrenzung zwischen Medizinprodukten und allgemeinen Verbraucherprodukten. Dieses Kapitel befasst sich mit Produkten, die einen medizinischen Zweck haben können oder auch nicht. Es ist wahrscheinlich das nützlichste Kapitel für Hersteller von Konsumgütern, die medizinische Versprechen machen oder machen wollen (z.B. biofunktionelle Kleidung). Beachten Sie, dass die in Anhang XVI der EU-MDR aufgeführten Produkte als Medizinprodukte gelten, auch wenn sie keine medizinische Zweckbestimmung haben.
- Kapitel 1.1.9: Andere Grenzbereiche für Medizinprodukte.
Der Teilbereich der IVDR-Qualifikation umfasst nur 3 Kapitel:
- Kapitel 2.1.1: Abgrenzung zwischen IVDs und Medizinprodukten. Es sei darauf hingewiesen, dass noch unter der früheren Richtlinie 98/79/EG (IVDD) eine Leitlinie, MEDDEV 2.14/1, herausgegeben wurde mit einer soliden regulatorischen Grundlage für die Abgrenzung zwischen IVDs und Nicht-IVDs, die in Ermangelung von IVDR-spezifischen MDCG-Leitlinien und den in diesem Handbuch beschriebenen Grenzfällen weiterhin von Nutzen sein kann.
- Kapitel 2.1.2: Abgrenzung zwischen IVDs und allgemeinen Laborgeräten. Es wurde auch ein separater MDCG-Leitfaden veröffentlicht (siehe unten).
- Kapitel 2.1.3: Andere IVD-Grenzfälle.
Das neue Handbuch enthält auch Abschnitte zu Klassifizierungsfragen, die gemäss den EU-MDR- und IVDR-Klassifizierungsregeln strukturiert sind.
Die Koordinierungsgruppe Medizinprodukte hat zudem spezifische Leitfäden veröffentlicht, die in Verbindung mit dem neuen Handbuch zur Abgrenzung und Klassifizierung gelesen werden müssen, wenn sie Ihr Produkt betreffen:
- Die im Oktober 2019 veröffentlichte MDCG 2019-11 enthält Leitlinien zur Qualifizierung und Klassifizierung von Software für Medizinprodukte (MDSW). Dies betrifft sowohl Software, die in ein Hardware-Medizinprodukt eingebettet ist, als auch eigenständige Software.
- Die im April 2022 veröffentlichte MDCG 2022-5 ist ein EU-MDR-spezifisches Leitliniendokument zu den Grenzfällen zwischen Medizinprodukten und Arzneimitteln.
Darüber hinaus können auch die MDCG-Leitlinien zu den Klassifizierungsregeln für Medizinprodukte eine nützliche Quelle für Stellungnahmen der Regulierungsbehörden sein, da sie zahlreiche Beispiele enthalten. Diese sind: MDCG 2021-24 für Medizinprodukte und MDCG 2020-16 für IVDs.

Nutzen Sie die verfügbaren Instrumente, um die Unsicherheiten bei der Qualifizierung von Boderline-Produkten zu bewältigen.
Kurz gesagt: Wenn Sie den Verdacht haben, dass Ihr Produkt ein Medizinprodukt sein könnte, raten wir Ihnen, den rechtlichen Rahmen sowie die oben erwähnten offiziellen Leitlinien und sogar die Urteile des EuGH zu konsultieren, die in der Datenbank InfoCuria Rechtssprechung öffentlich zugänglich sind.
Wenn Sie weiterhin Zweifel haben und der Meinung sind, dass Ihr Produkt ein Medizinprodukt der niedrigsten Risikoklasse sein könnte, können Sie bei Ihrer zuständigen nationalen Behörde um Unterstützung bitten. Falls diese keine Auskunft geben wollen, können Sie sich gerne bei uns melden. Wenn eine höhere Risikoklasse in Frage kommt, wenden Sie sich für Unterstützung als erstes an eine Benannte Stelle.
Mehr zur Klassifizierung von Medizinprodukten finden Sie hier.
Wie Decomplix helfen kann
Decomplix bietet Ihnen die Qualifizierung und Klassifizierung Ihres Produkts an oder eine weitergehende Beurteilung Ihrer Situation sowie einen Fahrplan für die Erlangung einer CE-Kennzeichnung. Hier finden Sie mehr zu unseren Dienstleistungen.



