
Sprachliche Anforderungen bei der “Kennzeichnung” von Medizinprodukten gemäss EU-MDR und IVDR
Vorneweg: Die sprachlichen Anforderungen der EU-MDR und IVDR beschränken sich nicht auf die “Kennzeichnung”. Sie gelten auch für verschiedene andere regulierte Dokumente wie EU-Konformitätserklärungen, amtliche Erklärungen, die technische Dokumentation, die der Konformitätsbewertung durch Benannte Stellen unterliegt, oder Sicherheitsmitteilungen im Feld.
Ersetzt die Version vom 17.06.2020
Wichtigste Erkenntnisse
- Der Begriff “Kennzeichnung” wird weit ausgelegt. Die sprachlichen Anforderungen betreffen nicht nur die direkte Kennzeichnung von Produkten, Verpackungsetiketten und Gebrauchsanweisungen, sondern auch Implantatkarten, SSCP/SSP oder die grafische Benutzeroberfläche (GUI) von Software und Marketingmaterialien.
- Die Anforderungen an die Kennzeichnungssprache liegen im Ermessen der EU-Mitgliedstaaten, Islands, Liechtensteins, Norwegens und der Türkei.
- Die Hersteller müssen sich mit den sprachlichen Anforderungen in jedem Land vertraut machen, in dem sie ihre Produkte vermarkten wollen. Fehlen die erforderlichen Übersetzungen der Kennzeichnung, so wird ein Produkt unrechtmässig in Verkehr gebracht.
Inhalt:
Welche Informationen sind von den sprachlichen Anforderungen der EU-MDR/IVDR betroffen? Und was ist eigentlich eine Kennzeichnung?
Der Begriff “Kennzeichnung” sollte für regulatorische Zwecke vermieden werden, da er verwirrend ist. “Kennzeichnung” wird in der Verordnungen (EU) Nr. 2017/745 über Medizinprodukte (EU-MDR) und 2017/746 über In-vitro-Diagnostika (IVDR) mehrfach verwendet und bezieht sich meist auf Verpackungsetiketten. Es gibt jedoch keine rechtliche Definition von “Kennzeichnung”.
Die neue Fassung der ISO 18113-1 zur IVD-Kennzeichnung bezieht sich auf die Definition der “Kennzeichnung” in EN ISO 13485:2016, die gleichbedeutend ist mit “vom Hersteller bereitgestellte Informationen” und Folgendes umfasst: Etiketten und Gebrauchsanweisung sowie alle Informationen zur Produktidentifizierung, Zweckbestimmung, technischen Beschreibung oder ordnungsgemässen Verwendung. Dieses Konzept entspricht der Empfehlung des International Medical Device Regulatory Forum (IMDRF) in seinen Kennzeichnungsgrundsätzen, IMDRF GRRP-WG/N52.
“Vom Hersteller bereitgestellte Informationen” ist in der Tat ein besserer Begriff als “Kennzeichnung”. Er entspricht dem, was in Kapitel III der grundlegenden Sicherheits- und Leistungsanforderungen gefordert wird (Anhang I EU-MDR und IVDR). Er umfasst die Informationen, die zur Identifizierung des Produkts und Herstellers erforderlich sind, sowie alle Sicherheits- und Leistungsinformationen, die der Hersteller mit dem Produkt liefern muss:
- Auf dem Produkt selbst (d.h. direkte Kennzeichnung, einschliesslich Kennzeichnung auf der Software),
- Auf den Verpackungsetiketten,
- Auf der Gebrauchsanweisung, unabhängig von der Form des Datenträgers (d.h. Papier- oder elektronische Form), und
- Auf der Website des Herstellers.
Weitere Dokumente, die als “vom Hersteller bereitgestellte Informationen” gelten, sind:
Implantatkarten
Hersteller von implantierbaren Produkten müssen zusammen mit dem implantierten Produkt eine Implantatkarte liefern (Art. 18 EU-MDR). Einige Produkte sind davon ausgenommen, z.B. Nahtmaterial, Klammern, Zahnfüllungen, Zahnspangen, Zahnkronen, Schrauben, Keile, Platten, Drähte, Stifte, Clips und Stecker. Darüber hinaus muss der Hersteller in der Gebrauchsanweisung Anleitungen für das Medizinpersonal geben, das die Implantatkarte ausfüllen muss.
Die Informationen auf der Implantatkarte müssen in der vom betreffenden Mitgliedstaat geforderten Sprache angegeben werden (Art. 18 Abs. 1 EU-MDR). Um Sprache und damit Übersetzungen zu vermeiden, empfiehlt der Leitfaden MDCG 2019-8 die Verwendung von anerkannten Symbolen. Es gibt jedoch kein Symbol für “Produkttyp”. Diese Information muss in der erforderlichen nationalen Sprache angegeben werden. Eine unvollständige Liste von Begriffen für den “Produkttyp” in englischer Sprache findet sich im Leitfaden MDCG 2021-11. Der Hersteller ist für deren korrekte Übersetzung verantwortlich.
Kurzbericht über Sicherheit und klinische Leistung (SSCP) / Kurzbericht über Sicherheit und Leistung (SSP)
Der SSCP, der für alle implantierbaren Produkte und Produkte der Klasse III vorgeschrieben ist (Art. 32 EU-MDR), und der SSP, der für alle IVD der Klassen C und D vorgeschrieben ist (Art. 29 IVDR), werden auch als “Kennzeichnung” bezeichnet. Sie liefern dem Endanwender oder dem Patienten wichtige Informationen über das Produkt. Daher müssen SSCP und SSP so verfasst sein, dass sie für den entsprechenden Leser verständlich sind.
MDCG 2019-9 empfiehlt Übersetzungen des SSCP analog zu den Anforderungen an die Gebrauchsanweisung. Demnach soll der für Patienten bestimmte Inhalt des SSCP in allen Sprachen bereitgestellt werden, die für Gebrauchsinformationen in den betreffenden Mitgliedstaaten vorgeschrieben sind. Eine englische Übersetzung muss immer bereitgestellt werden, auch wenn Englisch nicht zu den geforderten Sprachen gehört, da dies die gängigste Sprache in wissenschaftlichen Veröffentlichungen ist und vom Gesundheitspersonal weitgehend verstanden wird.
MDCG 2019-9 schreibt zudem vor, dass ein SSCP-Dokument pro Sprache erstellt wird und dass die von der Benannten Stelle validierte Sprache angegeben wird. In Analogie dazu könnte die gleiche Anforderung für den SSP gelten.
Wie sieht es mit der Sprache der grafischen Benutzeroberfläche in Medizinproduktesoftware aus?
Viele EWR-Länder haben keine spezifischen Anforderungen an die Sprache der grafischen Benutzeroberfläche (engl. graphic user interface, GUI), da das GUI als Bestandteil des Produkts und nicht als Kennzeichnung angesehen werden kann. Allerdings kann es bei standalone Software (einschliesslich mobiler Apps) schwierig sein, die Gebrauchsanweisung eindeutig von den Softwarefunktionen zu trennen, da beide auf dem Produkt selbst, d.h. auf dem GUI der Software, stehen. Es ist daher u.U. einfacher, das gesamte GUI als Kennzeichnung zu betrachten und es für die Länder, die eine Kennzeichnung in der Landessprache verlangen, vollständig übersetzen zu lassen.
Wenn das GUI als Komponente betrachtet wird, muss der Hersteller sicherstellen, dass es den grundlegenden Sicherheits- und Leistungsanforderungen entspricht (Anhang I EU-MDR/IVDR), insbesondere denjenigen, die sich auf Risiken im Zusammenhang mit Anwendungsfehlern beziehen. Möglich sind z.B. ein symbolbasiertes GUI, ein vollständig oder teilweise übersetztes GUI oder klare Erläuterungen des englischen GUI in der Gebrauchsanweisung für eine sichere und korrekte Anwendung. Der Hersteller sollte die gewählte Lösung begründen und sie in der Risikomanagementakte dokumentieren.
Einige Länder haben sehr eindeutige Positionen zur Sprache der grafischen Benutzeroberfläche.
- Dänemark: Auf der Website der dänischen Arzneimittelbehörde wird eingeräumt, dass es keine allgemeinen Anforderungen gibt und dass es über die Gebrauchsanweisung hinaus in der Verantwortung des Herstellers liegt, zu bestimmen, welche Informationen für die ordnungsgemässe und sichere Verwendung des Produkts erforderlich sind. Dabei wird implizit gefordert, dass diese Art von Informationen als “Kennzeichnung” betrachtet wird und somit der Übersetzung unterliegt. Sie weisen auch darauf hin, dass Schaltflächen, Tasten oder Anzeigen mit einzelnen Wörtern oder Begriffen wie “Laden”, “Enter” und “Seite nach unten” als Symbole betrachtet werden und von der Übersetzung auf der Software ausgenommen sind, aber in der Gebrauchsanweisung erklärt werden sollten.
- Ungarn: Auf der Website des Nationalen Instituts für Pharmazie und Ernährung OGYÉI finden sich auch Informationen zu Software-Übersetzungen. Im Allgemeinen muss die GUI in ungarischer Sprache verfügbar sein (mit Ausnahme von standardisierten Piktogrammen), unabhängig davon, ob das Produkt für den professionellen Gebrauch oder für Laien bestimmt ist. Was die Anzeigen betrifft, so können sie auf Englisch sein, wenn sie sehr einfach sind und die ungarische Gebrauchsanweisung die Screenshots erklärt. OVYÉI weist die Hersteller darauf hin, dass selbst bei professioneller Nutzung “it cannot be expected from service providers to have a certified and employer-accepted language proficiency in any language other than Hungarian”.
- Norwegen: Die Website der norwegischen Arzneimittelbehörde enthält ausführliche Leitlinien zur “Kennzeichnung” und schreibt ausdrücklich vor, dass die Software in norwegischer Sprache sein muss.
- Polen hat detaillierte gesetzliche Anforderungen an die Sprache des GUI. Das GUI von Software für Laien muss in Polnisch sein. Eine englische GUI ist jedoch zulässig wenn:
- Das Produkt nicht für die Verwendung in Situationen vorgesehen ist, die eine unmittelbare Gefahr für Gesundheit und Leben darstellen,
- alle Konzepte, Symbole und Befehle in der polnischen Gebrauchsanweisung erklärt werden,
- die englische Benutzeroberfläche keine Gefahr für den Benutzer darstellt und auf der Verpackung deutlich markiert ist. Software für den professionellen Einsatz kann ein englisches GUI haben, mit Ausnahme des für den Patienten bestimmten Teils des GUI, für den die vorherigen Bedingungen gelten.
- Rumänien hat per Gesetz GUI-Sprachanforderungen eingeführt. GUI für Nicht-Fachleute müssen in Rumänisch bereitgestellt werden. Englischsprachige GUI sind in begründeten Fällen nur für den professionellen Gebrauch und mit schriftlicher Zustimmung des Nutzers zulässig.
Wie sieht es mit Marketingmaterialien aus?
Marketing- oder Werbematerialien werden in der EU-MDR oder IVDR nicht als Teil der vom Hersteller bereitzustellenden Informationen erwähnt. Im Prinzip gelten hier die sprachlichen Anforderungen in den nationalen Rechtsvorschriften zur Übernahme der EU-MDR und der IVDR nicht.
Die Norm EN ISO 20417 über die vom Medizinproduktehersteller bereitzustellenden Informationen schliesst Werbematerialien (so genannte Marketinginformationen) ausdrücklich aus ihrem Anwendungsbereich aus. Sie weist aber darauf hin, dass einige Rechtsordnungen solche zusätzlichen Informationen als vom Hersteller bereitgestellte Informationen betrachten können, was beispielsweise in den USA der Fall ist. Dieser Ausschluss vom Anwendungsbereich wird wortwörtlich aus den IMDRF-Prinzipien für die Kennzeichnung von Medizinprodukten und IVD, IMDRF GRRP-WG/N52, übernommen
In den nationalen Rechtsvorschriften der EWR-Länder und der Türkei werden Marketingmaterialien nicht ausdrücklich als “vom Hersteller bereitgestellte Informationen” betrachtet (das gilt auch für Marketingmaterialien, die der Hersteller auf seiner Webseite bereitstellt). Die nationale Gesetzgebung über Werbung und Verkaufsförderung für Medizinprodukte kann jedoch zusätzliche sprachliche Anforderungen enthalten.
Welche Sprachen sollte der Hersteller für die Übersetzung der “Kennzeichnung” in Betracht ziehen?
Laut EU-MDR und IVDR muss der Hersteller dafür sorgen, “dass dem Produkt die Informationen gemäss Anhang I Abschnitt 23 in einer oder mehreren von dem Mitgliedstaat, in dem das Produkt dem Anwender oder Patienten zur Verfügung gestellt wird, festgelegten Amtssprache(n) der Union beiliegen.” (Art. 10 Abs. 11 EU-MDR, Art. 10 Abs. 10 IVDR)
Mit anderen Worten: Die EU-MDR und die IVDR geben keine Auskunft darüber, welche Sprachen für welche “Kennzeichnungselemente” erforderlich sind und welche Ausnahmen gelten. Der Hersteller muss sich in den nationalen Rechtsvorschriften der einzelnen Länder informieren, in denen er seine Produkte verkaufen will. Als Hilfeleistung hat die EU-Kommission auf einer eigens eingerichteten Webseite die folgenden Zusammenstellungen von Sprachanforderungen veröffentlicht:
Diese Zusammenstellungen bieten eine hilfreiche Grundlage, sie sind aber für einige Länder oder Aspekte nicht aktuell.
Der EWR umfasst die derzeit 27 Mitgliedstaaten der Europäischen Union (EU) sowie 3 der 4 Mitglieder der Europäischen Freihandelsassoziation (EFTA), d.h. Island, Liechtenstein und Norwegen. Die Schweiz ist zwar EFTA-, jedoch nicht EWR-Mitglied. Siehe auch Kapitel “Was ist mit der Schweiz?”.
Die folgende Tabelle zeigt die offiziellen Landessprachen im EWR und in der Türkei.
-
Land
Verhältnis zur EU
Offizielle Sprache(n)
Österreich
EU Mitglied
Deutsch
Belgien
EU Mitglied
Niederländisch, Französisch, und Deutsch
Bulgaria
EU Mitglied
Bulgarisch
Kroatien
EU Mitglied
Kroatisch
Cyprus
EU Mitglied
Griechisch und Türkisch
Tschechische Republik
EU Mitglied
Tschechisch
Dänemark
EU Mitglied
Dänisch
Estland
EU Mitglied
Estnisch
Finnland
EU Mitglied
Finnisch und Schwedisch
Frankreich
EU Mitglied
Französisch
Deutschland
EU Mitglied
Deutsch
Griechenland
EU Mitglied
Griechisch
Ungarn
EU Mitglied
Ungarisch
Island
EFTA Mitglied
Isländisch
Irland
EU Mitglied
Irisch und Englisch
Italien
EU Mitglied
Itälienisch
Lettland
EU Mitglied
Lettisch
Liechtenstein
EFTA Mitglied
Deutsch
Litauen
EU Mitglied
Litauisch
Luxemburg
EU Mitglied
Französisch, Deutsch und Luxemburgisch
Malta
EU Mitglied
Maltesisch und Englisch
Norwegen
EFTA Mitglied
Norwegisch
Niederlanden
EU Mitglied
Niederländisch
Polen
EU Mitglied
Polnisch
Portugal
EU Mitglied
Portugiesisch
Rumänien
EU Mitglied
Rumänisch
Slovakei
EU Mitglied
Slovakisch
Slowenien
EU Mitglied
Slowenisch
Spanien
EU Mitglied
Spansich
Schweden
EU Mitglied
Schwedisch
Türkei
Zollunion
Türkisch
Die sprachlichen Anforderungen im EWR sind eine Herausforderung. Einerseits gibt es 27 Sprachen, zusätzlich zum Englischen, wobei manche von ihnen nicht für die Kennzeichnung von Medizinprodukten vorgeschrieben sind, andererseits machen einige Länder Ausnahmen für die berufliche Verwendung geltend. Dies führt zu einem Flickenteppich von Sprachanforderungen, wie im nächsten Abschnitt beschrieben.
Wie setzen die EWR-Länder die sprachlichen Anforderungen bei der “Kennzeichnung” um?
Die sprachlichen Anforderungen bei der Kennzeichnung sind in den betreffenden nationalen Rechtsvorschriften der einzelnen EWR-Länder und der Türkei enthalten. Manchmal werden zusätzliche Informationen oder Stellungnahmen auf den Websites der zuständigen nationalen Behörden veröffentlicht. Die nationalen Rechtsvorschriften verweisen meist auf die EU-MDR und den Abschnitt über die vom Hersteller bereitzustellenden Informationen (Anhang I Kapitel III), dadurch gelten die Anforderungen implizit sowohl für Etiketten als auch für Gebrauchsanweisungen.
Unter den früheren Richtlinien (MDD, AIMDD, IVDD) war der Ansatz in Bezug auf die Kennzeichnungssprache sehr kategorisch. Die meisten Länder liessen nur die Landessprache zu und es gab sehr wenig Raum für Ausnahmen oder Englisch für den professionellen Gebrauch. Dies hat sich mit der EU-MDR/IVDR geändert.
Heute sind per Gesetz verschiedene Situationen möglich:
- Die Landessprache ist ausnahmslos obligatorisch.
Betroffene Länder: Bulgarien, Frankreich, Italien, Litauen, Portugal, Spanien, Tschechien und Ungarn - Die Landessprache ist vorgeschrieben, aber Teile der Kennzeichnung können in Englisch oder in anderen Sprachen geschrieben sein (in der Regel Informationen, die sich nicht auf die Sicherheit beziehen).
In der Praxis vereinfachen diese “Ausnahmen” die Übersetzungen nicht sehr, so dass diese Gruppe wie die vorherige ist. Betroffene Länder: Finnland und Deutschland. - Die Landessprache ist vorgeschrieben, aber Ausnahmen für den beruflichen Gebrauch sind möglich (Englisch oder andere Sprachen), unter Bedingungen und vorbehältlich einer vorherigen Genehmigung, die zeitlich begrenzt sein kann.
Betroffene Länder: Dänemark, Griechenland, Norwegen, Rumänien, Schweden und die Türkei. - Die Landessprache ist vorgeschrieben, aber Ausnahmen sind zulässig für den beruflichen Gebrauch (Englisch oder andere Sprachen), in einigen Fällen unter bestimmten Bedingungen, aber ohne vorherige Genehmigung.
Betroffene Länder: Österreich, Belgien, Kroatien, Zypern, Estland, Island, Lettland, Liechtenstein, Luxemburg, Niederlande, Polen, Slowakei und Slowenien. - Englisch wird ohne Einschränkung akzeptiert (d.h. sowohl für Laien als auch für Fachleute).
Betroffene Länder: Irland und Malta.
Darüber hinaus ist Nordirland aufgrund der Post-BREXIT-Abkommen zwischen dem Vereinigten Königreich und der EU ein Gebiet des Vereinigten Königreichs, in dem die EU-MDR und die IVDR gelten und in dem nur Englisch für die Kennzeichnung zulässig ist.
Diese Fälle sind in der folgenden EWR-Karte allgemein dargestellt (d.h. ohne die Türkei, aber einschliesslich der Nicht-EWR-Schweiz).
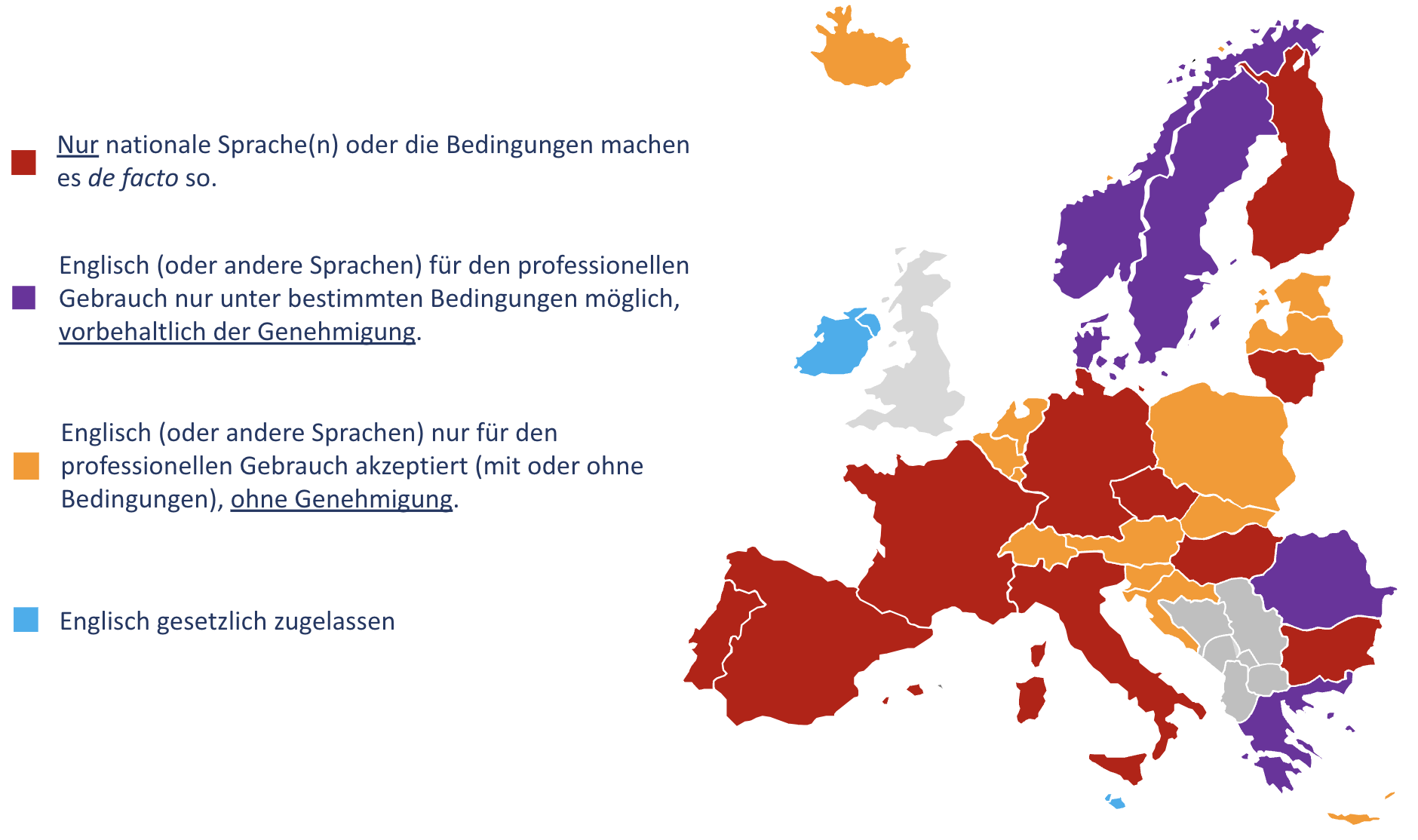 Hersteller von “Legacy”-Produkten (die noch nach den früheren Richtlinien CE-gekennzeichnet sind) müssen jedoch beachten, dass die oben beschriebenen Situationen den nationalen Gesetzgebungen entsprechen, die wiederum der EU-MDR/IVDR angepasst wurden. Diese gelten daher u.U. nicht für “Legacy”-Produkte.
Hersteller von “Legacy”-Produkten (die noch nach den früheren Richtlinien CE-gekennzeichnet sind) müssen jedoch beachten, dass die oben beschriebenen Situationen den nationalen Gesetzgebungen entsprechen, die wiederum der EU-MDR/IVDR angepasst wurden. Diese gelten daher u.U. nicht für “Legacy”-Produkte.
Was ist mit der Schweiz?
Seit der Unterbrechung des Abkommens über die gegenseitige Anerkennung (MRA) zwischen der Schweiz und der EU wird die Schweiz als Drittland betrachtet und die EU-MDR/IVDR gelten nicht direkt. Die Schweiz hat jedoch beschlossen, die Schweizerische Medizinprodukteverordnung (MepV) und die Schweizerische In-vitro-Diagnostika-Verordnung (IvDV) der EU-MDR und IVDR anzupassen und denselben Grundsätzen für die “Kennzeichnung” zu folgen.
Die Kennzeichnung von Medizinprodukten müssen in allen drei Amtssprachen der Schweiz, d.h. in Deutsch, Französisch und Italienisch, verfügbar sein (Art. 16 Abs. 2 MepV, Art. 15 Abs. 2 IvDV). Anstelle des Klartextes können durch technische Normen festgelegte Symbole verwendet werden. Es ist möglich, die Kennzeichnung in weniger als den drei Schweizer Amtssprachen oder nur in Englisch vorzunehmen (Art. 16 Abs. 3 MepV, Art. 15 Abs. 3 IvDV), sofern:
- “das Produkt ausschliesslich Gesundheitsfachpersonen abgegeben wird oder es sich beim Produkt um eine Sonderanfertigung oder um ein Produkt handelt, das ausschliesslich in einer Gesundheitseinrichtung verwendet wird;
- die Anwenderin oder der Anwender die notwendigen fachlichen und sprachlichen Voraussetzungen mitbringt und damit einverstanden ist;
- der Schutz von Patientinnen und Patienten, von Anwenderinnen und Anwendern sowie Dritter gewährleistet ist; und
- die wirksame und leistungsbezogene Anwendung nicht gefährdet wird.”
Auf Verlangen des Benutzers müssen Informationen zusätzlich in einer der drei Schweizer Amtssprachen bereitgestellt werden (Art. 16 Abs. 4 MepV, Art. 15 Abs. 4 IvDV).
In Liechtenstein gelten sehr ähnliche Bedingungen (wenn auch nur für die deutsche Sprache), da beide Länder durch den Zollvertrag, RS 0.631.112.514, verbunden sind. Anhang I des Zollvertrags (verfügbar im liechtensteinischen Gesetzblatt Nr. 280) gibt an, dass die Schweizerische MepV und IvDV in Liechtenstein gelten, wobei nur einige wenige Artikel ausgeschlossen sind.
Was bedeuten die sprachlichen Anforderungen bei der “Kennzeichnung” für die Hersteller?
Medizinproduktehersteller (einschliesslich IVD) können die sprachlichen Anforderungen der einzelnen Länder, in denen sie ihre Produkte vermarkten wollen, nicht ignorieren. Fehlt die geforderte “Kennzeichnungssprache”, wird ein Produkt nicht ordnungsgemäss auf den Markt gebracht. Es ist für Hersteller dringend erforderlich, die aktuellen Sprachanforderungen in den Zielländern des EWR und der Türkei zu berücksichtigen.
Übersetzungen für Produktkennzeichnungen können teilweise umgangen werden, indem international anerkannte Symbole aus harmonisierten Normen oder gemeinsamen Spezifikationen verwendet werden. Die harmonisierte Norm EN ISO 15223-1:2021 ist die Hauptquelle für Symbole im Rahmen der EU-MDR und IVDR. Werden nicht-anerkannte Symbole oder Kennfarben benutzt, so müssen diese in der Produktdokumentation, im Wesentlichen in der Gebrauchsanweisung, erläutert werden. Das bedeutet, dass sie wiederum der Übersetzung unterliegen.
Letztendlich bedeutet dies, dass das Qualitätsmanagementsystem des Herstellers ein Übersetzungsverfahren enthalten muss, in dem beschrieben wird, wer für die Durchführung von Übersetzungen qualifiziert ist, wie die Genauigkeit der Übersetzungen überprüft werden soll (z.B. durch Rückübersetzungen) und wie die Übersetzungen auf dem neuesten Stand gehalten werden, wenn sich die Originalversion ändert. Wenn ein Medizinprodukt oder ein IVD eine Software-Benutzeroberfläche enthält, muss der Software-Design- und -Entwicklungsprozess bereits die Anforderungen an die Sprache der Benutzeroberfläche mitbedenken. Dabei müssen auch Risikomanagement- und Usability-Engineering-Aspekte berücksichtigt werden.
Dies bedeutet, dass die internen Produktfreigabeprozesse des Herstellers angepasst werden. Die Produkte dürfen nur dann in die jeweiligen Länder geliefert werden, wenn sie mit der erforderlichen übersetzten Kennzeichnung versehen sind, oder wenn gewährleistet ist, dass die Benutzer die übersetzte Kennzeichnung auf Anfrage erhalten.

Medizinproduktehersteller können die Anforderungen der einzelnen Länder an die Kennzeichnungssprache nicht ignorieren.
Welche Verantwortung müssen Importeure und Händler übernehmen?
Importeure und Händler sind indirekt für die Einhaltung der Vorschriften bei der Kennzeichnungssprache verantwortlich, da sie die Kennzeichnung überprüfen müssen, bevor sie das Produkt auf dem Markt bereitstellen (Art. 13 Abs. 2 bzw. Art. 14 Abs. 2 EU-MDR/IVDR).
Wenn ein Importeur oder Händler eine länderspezifische Umetikettierung eines Medizinprodukts vornimmt, die auch die Übersetzung der Gebrauchsanweisung und anderer Unterlagen umfassen kann, gilt Artikel 16 der EU-MDR/IVDR. Wenn der Importeur/Händler die Übersetzung oder das Umpacken/Umetikettieren selbst vornimmt, gilt Folgendes:
- Die durchgeführte Tätigkeit (d.h. Übersetzung oder Umpacken/Umetikettieren) wie auch sein Name und seine Anschrift sind auf dem Produkt oder der Verpackung oder den dem Produkt beigefügten Unterlagen anzugeben (Art. 16 Abs. 3).
- Der Importeur/Händler verfügt über ein Qualitätsmanagementsystem (QMS), das sicherstellt, dass die Übersetzungen korrekt und auf dem neuesten Stand sind, dass die damit verbundenen Umetikettierungs- und/oder Umverpackungsaktivitäten keine Auswirkungen auf den ursprünglichen Zustand des Produkts haben, dass die neue Verpackung von angemessener Qualität ist und dass der Importeur/Händler über alle vom Hersteller ergriffenen Korrekturmassnahmen in Bezug auf das betreffende Produkt informiert wird (Art. 16 Abs. 3). Beachten Sie, dass ein solches QMS von einer Benannten Stelle zertifiziert sein muss, wie im Leitfaden MDCG 2021-23 näher beschrieben.
- Der Hersteller und die betroffenen zuständigen nationalen Behörden müssen über das umetikettierte/umverpackte Produkt informiert werden, und zwar 28 Tage vor der Bereitstellung auf dem Markt (Art. 16 Abs. 4).
- Der Importeur/Händler legt der zuständigen Behörde innerhalb der gleichen Frist von 28 Tagen die von einer Benannten Stelle ausgestellte Bescheinigung vor, in der bestätigt wird, dass das bestehende QMS den Anforderungen entspricht (Art. 16 Abs. 4).
- Der Importeur/Händler muss in der Lage sein, der zuständigen Behörde auf Anfrage Muster des umetikettierten/umverpackten Produkts (inkl. Übersetzungen) vorzulegen (Art. 16 Abs. 4).
Anders sieht es aus, wenn das Umverpacken oder Umetikettieren (inkl. Übersetzungen) im Auftrag und unter der Kontrolle des Herstellers erfolgt. Wie im Leitfaden MDCG 2021-26 deutlich erklärt, gelten Art. 16 Abs. 2, 3 und 4 in diesem Fall nicht. Das Umverpacken oder Umetikettieren, das im Auftrag des Herstellers erfolgt, gilt lediglich als Tätigkeit im Unterauftrag und muss entsprechend gehandhabt werden.
Spezifische Details zu den Auswirkungen von Übersetzungen auf Schweizer Importeure finden Sie in unserem diesbezüglichen Blog-Artikel.
Wie Decomplix helfen kann
Unser Team berät und unterstützt Sie in allen regulatorischen Belangen und gibt Ihnen Auskunft bei speziellen Fragen rund um die EU-MDR und die IVDR sowie zu den Sprachregelungen. Wir beobachten die Entwicklungen in allen Mitgliedstaaten und unser Expertenteam steht Ihnen jederzeit zur Verfügung.
Zögern Sie nicht, uns zu kontaktieren.
Weiterlesen:
- Schweizer Importeure von Medizinprodukten – rechtliche Anforderungen
- Welche Anforderungen der EU-MDR gelten für Händler von Medizinprodukten?
- Anforderungen an Medizinprodukte der Klasse I für Hersteller gemäss EU-MDR
- Qualitätsmanagement-System nach ISO 13485 zertifizieren
- Harmonisierte Normen für Medizinprodukte und IVDs verstehen



